Articles
- Page Path
- HOME > Endocrinol Metab > Volume 37(4); 2022 > Article
-
Review ArticleAdrenal Gland Long-Term Outcomes of Congenital Adrenal Hyperplasia
 Keypoint
Keypoint
Due to its complex nature, congenital adrenal hyperplasia (CAH) and its treatment may result in several negative long-term outcomes. New treatments and improved monitoring during follow-up may further improve patients’ long-term health. Regular monitoring of various risk factors and complications is essential to identify problems and institute support and treatment at an early stage. This review discusses the different long-term outcomes of CAH. -
Anna Nordenström1,2
 , Svetlana Lajic1,2, Henrik Falhammar3,4
, Svetlana Lajic1,2, Henrik Falhammar3,4 -
Endocrinology and Metabolism 2022;37(4):587-598.
DOI: https://doi.org/10.3803/EnM.2022.1528
Published online: July 8, 2022
1Department of Women’s and Children’s Health, Karolinska Institute, Stockholm, Sweden
2Pediatric Endocrinology Unit, Karolinska University Hospital, Stockholm, Sweden
3Department of Molecular Medicine and Surgery, Karolinska Institute, Stockholm, Sweden
4Department of Endocrinology, Karolinska University Hospital, Stockholm, Sweden
- Corresponding author: Henrik Falhammar. Department of Endocrinology, Karolinska University Hospital, SE-171 76 Stockholm, Sweden Tel: +46-851776411, Fax: +46-851773096, E-mail: henrik.falhammar@ki.se
Copyright © 2022 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- A plethora of negative long-term outcomes have been associated with congenital adrenal hyperplasia (CAH). The causes are multiple and involve supra-physiological gluco- and mineralocorticoid replacement, excess adrenal androgens both intrauterine and postnatal, elevated steroid precursor and adrenocorticotropic hormone levels, living with a congenital condition as well as the proximity of the cytochrome P450 family 21 subfamily A member 2 (CYP21A2) gene to other genes. This review aims to discuss the different long-term outcomes of CAH.
- Congenital adrenal hyperplasia (CAH) is a rare group of autosomal recessive disorders, the most common being 21-hydroxylase deficiency (21OHD) affecting up to 99% of all CAH cases [1-3]. More than 95% of all cases with 21OHD are due to 10 common pathogenic variants in the cytochrome P450 family 21 subfamily A member 2 (CYP21A2) gene which are due to intergenic recombination events with the CYP21A1P pseudogene [3,4]. The impairment of the 21-hydroxylase enzyme results in variable degree of cortisol and aldosterone deficiency and a concomitant increased production of adrenal androgens [5-7]. CAH can be divided into three phenotypes, i.e., salt-wasting (SW), simple virilizing (SV), and non-classic (NC) CAH, the first two are referred to as classic CAH [8]. There is a good genotype-phenotype correlation [9] and analyzing the steroid precursor 17-hydroxyprogesterone (17OHP) levels is the gold standard to diagnose 21OHD [10]. Neonatal screening for 21OHD is today available in many parts of the world and most cases of classic CAH (incidence 1:10,000–1:20,000) are therefore detected during the first week of life [5,11]. Girls with classic CAH present with varying degrees of virilization of external genitalia. If untreated, both girls and boys with SW CAH may develop life threatening neonatal SW crisis. Males, if not subjected to neonatal screening, are diagnosed due to SW crisis or precocious puberty [3]. Thus, neonatal screening is of particular benefit in males [1,11,12]. However, most cases with NC CAH are missed by neonatal screening [9]. The presenting symptoms of androgen excess in patients with NC CAH are milder than in classic CAH and the diagnosis is usually established later in childhood or in young adults [13].
- Prior to the introduction of glucocorticoid replacement in the 1950s survival was not possible in cases with the most severe phenotype [1,8]. After the introduction of glucocorticoid and later mineralocorticoid treatment (mineralocorticoid used if needed) this has been the standard treatment [3,8,14]. The balancing act between symptoms of hyperandrogenism and hypercortisolism is, however, complex. Unfortunately, supra-physiological glucocorticoid doses are typically required for sufficient hypothalamic–pituitary–adrenal axis suppression which will result in unfavorable long-term outcomes [3,8,15,16]. The more long-acting the glucocorticoid is the more it seems to affect different outcomes [17]. However, there are no prospective randomized studies comparing various glucocorticoid regimens to investigate glucocorticoid and androgen effects on long-term outcomes [18]. Moreover, some long-term outcomes are a result of the high androgen exposure during fetal life or due to the proximity of the CYP21A2 gene to other genes.
- The aim of this review is to discuss different long-term outcomes in patients with CAH.
INTRODUCTION
- Both over- and undertreatment with glucocorticoid results in compromised final height, in males and females with CAH [3]. Meta-analyses have shown that patients lose about 1 standard deviation scores in final height corrected for parental height [19,20]. The achieved final height was somewhat better in more recent studies and mineralocorticoid supplementation and early diagnosis seem to be beneficial [20-22].
GROWTH
- Most studies have found an elevated cardiovascular and metabolic risk in patients with CAH [2,23-33]. In a Swedish registry-based study cardiovascular disease (odds ratio [OR], 2.7), hypertension, hyperlipidemia, atrial fibrillation, venous thromboembolism, obesity, diabetes (mainly type 2) and obstructive sleep disorder were increased in patients with CAH compared to matched controls [27]. Women and patients with the NC phenotype had a generally higher morbidity risk. In a Korean epidemiological study, cardiovascular disease (OR, 1.4), stroke, diabetes, hyperlipidemia and hypertension were more prevalent in patients with CAH compared to matched controls [33]. Venous thromboembolism showed a tendency to be more prevalent as well (P=0.055).
- Body mass index (BMI) is often high in patients with CAH [2,23,26,30-32,34]. However, this could partly reflect an increase in lean mass in women with CAH due to high androgen exposure during life [23]. In a meta-analysis of both children and adults with CAH, patients had elevated homeostatic model assessment for insulin resistance levels compared to controls [29]. Others found that gestational diabetes, a prediabetes condition, was more likely in pregnant women with CAH than in matched controls [35-37]; however, when adjusted for, among other things obesity, it was no longer significantly increased [37]. The outcome from studies on blood pressure varies, with some studies showing elevated blood pressure in patients with CAH while others do not confirm this [2,23,25,26,28,31,32,38, 39]. Patients with CAH had higher blood pressure in a meta-analysis including both patients with CAH and matched controls [29]. Especially in children, the risk for hypertension seems elevated, and the relative risk (RR) compared to matched controls appears to decrease with age, especially in females with CAH [27,33]. Others have shown that younger children have less hypertension than adolescents with CAH [10]. However, the highest risk was identified in children below the age of 2 years [28] suggesting that high fludrocortisone doses can cause hypertension in young children while high BMI could be the main cause later in life [10].
- If the lipid profile is at a disadvantage in patients with CAH is under debate [2,23,25-28,30,38] and in the previously mentioned meta-analysis similar lipid levels were reported in patients with CAH and matched controls [29].
- Elevated heart rate is associated with cardiovascular death, particular in men, independent of other risk factors [40,41]. Just a few more beats per minute (bpm), even within normal range, may increase the risk of cardiovascular events [41]. However, only occasional studies have reported elevated heart rates in subjects with CAH. One study demonstrated higher mean 24-hour heart rate in adults with CAH than in BMI-matched controls (by 3 bpm) [39]. In adult males with CAH only those aged 30 years or older had elevated 24-hour heart rate compared to matched controls (by 13 during the day and 27 bpm during the night, respectively) [25], although the effect may be due to obesity in the patient group.
- Carotid intima-media thickening (CIMT) measured with ultrasound, is an easy, non-invasive method to find subclinical atherosclerosis [42]. CIMT has been reported to be increased in both children and adults with CAH compared to controls [29,43,44]. The difference was less evident in children and adolescents than in adults with CAH [29].
CARDIOMETABOLIC DISORDERS
- Bone mineral density (BMD) has been reported to be anything from increased to decreased but most studies have found low BMD in at least one of the measured locations in both sexes and across all age groups [2,26,45-53]. A recent meta-analysis including 254 patients with CAH and 344 matched controls found a lower BMD in CAH in total body, lumbar spine and femoral neck, respectively [54]. Present or accumulated glucocorticoid dosages were negatively correlated to BMD [50-52,55]. Androgen excess seemed to improve BMD in both adult females and young patients with CAH [50,53]. Furthermore, patients with the classic phenotype had worse BMD compared to those with NC CAH [49]. Even though many studies have evaluated BMD in CAH the association between BMD and fractures is poor, especially in glucocorticoid-induced osteoporosis [56].
- Fracture risk in CAH has been less thoroughly studied. In a meta-analysis, including individuals with glucocorticoid replacement therapy, treatment increased the fracture risk [57]. Recent epidemiological studies on the presence of fractures in patients with CAH have been reported [33,58]. A Swedish study identified more fractures in the patient group already at a young age compared to the general population (OR, 1.61; mean age of the CAH group 29.8 years) [58]. However, in a Korean study, more fractures were only found in patients aged 40 years and older (OR, 1.4) [33]. In the Swedish study, only those with classic CAH had an increased fracture rate in contrast to individuals with NC CAH who had a fracture rate similar to matched controls [58]. This is in accordance with the findings of a United States study where patients with NC CAH had a lower fracture rate than patients with classic CAH [49]. However, the increased fracture risk in classic CAH may be something of the past since patients with CAH born after the introduction of neonatal screening for CAH had a similar fracture rate compared to matched controls in contrast to those born before the era of neonatal screening [58]. It could be speculated that an early diagnosis and modern management of CAH are superior in reducing fracture occurrence than prolonged androgen exposure on BMD.
BONE HEALTH
- Only few studies have investigated psychiatric co-morbidity in CAH [30,33,59-61]. The OR of having a psychiatric disorder was found to be between 1.5 and 1.9 compared to controls [33, 59,60], with increased depression [61], alcohol misuse [59,60] and suicidality [30,59]. Reaction to severe stress, and adjustment disorders were more prevalent in females with CAH than in matched controls [60].
PSYCHIATRIC DISEASES
- The CYP21A2 gene is located in a highly immunologically active region; hence, it could be speculated that 21OHD would have an effect on autoimmune disorders. On the other hand, the usage of glucocortiocoid replacement required in most patients with 21OHD [14,16], could be assumed to have an immunomodulating effect [62]. In a study of 145 patients with 21OHD, 3.4% had a concurrent autoimmune disorder [63], while in a pan-European study of disorders of sexual development, 22.2% of 222, mostly female patients with CAH, had a concurrent autoimmune disorder [30].
- The frequency of autoimmune disorders was demonstrated to be increased in individuals with 21OHD compared to age- and sex-matched controls and increased with age [64]. In this register-based study of 714 patients with 21OHD and 71,400 controls, 7.4% of patients with 21OHD had at least one autoimmune disorder compared to 5.1% of controls (RR, 1.47). Males with 21OHD had a higher risk than females (RR, 1.64 vs. 1.37), mainly due to the fact that autoimmune disorders are more common in females in general. The most prevalent disease was autoimmune thyroid disease which was increased in both sexes.
AUTOIMMUNE DISORDERS
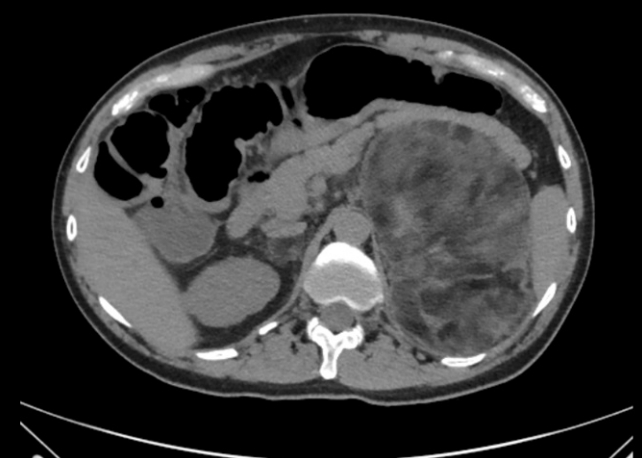
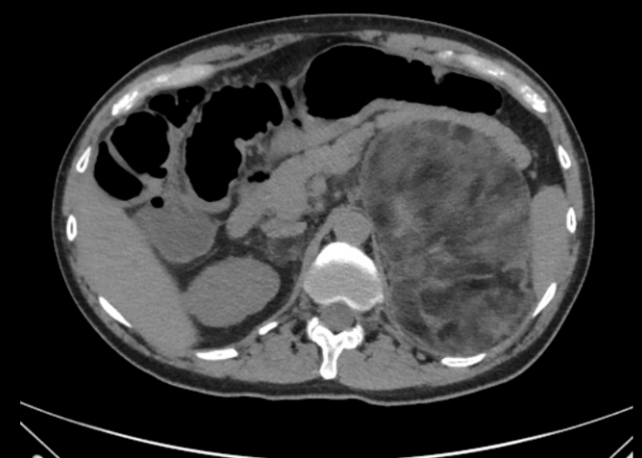
- In patients with non-functional adrenal incidentalomas, especially bilateral tumors or large adrenal myelolipomas, undiagnosed CAH should be considered [65-68]. In a meta-analysis 0.8% of all adrenal incidentalomas was due to a previously undiagnosed genetically confirmed CAH [69]. In another metaanalysis of patients with CAH a quarter had an adrenal tumor of which a quarter was a myelolipoma [70]. Nearly all of the patients with CAH that had a myelolipoma had a late diagnosis or poorly managed CAH [70]. Fig. 1 illustrates bilateral myeolipomas in a patient with CAH. The 17OHP levels correlate with adrenal tumor size and to avoid over-diagnosis of NC CAH genotyping should be performed [66,69]. Adrenocorticotropic hormone (ACTH) is considered the driver of adrenal hyperplasia and later adrenal tumor development [70,71].
- Adrenal rest tumors are common in the testicles of males with CAH and are called testicular adrenal rest tumors (TARTs) [72]. The prevalence of TARTs is around 40% [72], with the highest prevalence reported being 86% [73]. Most TARTs are bilateral [38,73,74]. The prevalence differs depending on the age, severity and probably the ultrasonography equipment if ultrasound of the testicles is performed. Ultrasonography is the preferred method since it is as sensitive as magnetic resonance imaging (MRI) but cheaper and more accessible [75]. Ovarian rest tumors are on the other hand rarely reported [76], possibly due to their location in the abdominal cavity where the use of ultrasonography is not optimal. MRI, or even better positron emission tomography/computer tomography can be used to detect both ovarian and retroperitoneal adrenal rest tumors [77]. ACTH has been proposed to be the most important stimulant of adrenal rest tumors [72]. Thus, intensifying glucocorticoid treatment may reduce the size of adrenal rest tumors in early stages [76] while bilateral adrenalectomy may increase the size [77,78]. It should be noted, though, that there is not always an association between poor control and adrenal rest tumors [32].
TUMORS
- Mortality has not been studied extensively in CAH. In countries without neonatal screening more females than males with SW CAH were diagnosed [12]. However, not only males with SW CAH died undiagnosed in the neonatal period, as was shown by the finding that when neonatal screening was introduced more individuals with SW CAH were found in both sexes [1]. Even when the patients had been diagnosed with CAH the mortality rate was increased with a hazard ratio of 1.6 to 5.17 [33,61,79]. The main cause of death was adrenal crisis, followed by cardiovascular disease (mainly stroke) [79]. However, half of the cases reported as cardiovascular deaths had concomitant severe infection which means that adrenal crisis may be contributing to the majority of the deaths. Moreover, in a survey sent out to German pediatric endocrinologists regarding causes of deaths among their children with CAH, all or almost all deaths were due to adrenal crisis and half had deceased at home [80]. Thus, adrenal crisis can be easily missed both by patients and health professionals inexperienced in adrenal insufficiency [81,82]. Especially in young children and old adults the symptoms can be difficult to interpret [83,84]. However, adolescents and young adults seem to be particularly at risk of adrenal crisis [85].
MORTALITY
- Gonadal function has been extensively studied in both sexes in CAH [76]. There are similarities in the gonadal dysfunction between the sexes but also some major discrepancies. In adolescent girls and women with CAH gonadal dysfunction can cause abnormal pubertal development and irregular menses including amenorrhea [76]. The age of pubertal onset and menarche in girls with CAH is similar to controls [35,86]. Since elevated androgen levels and steroid precursors can result in irregular menstruations, the presence of regular menstruations, and especially ovulation, suggest good hormonal control. However, irregular menstruations are also common in the general population and women with CAH may in fact not have a higher frequency [35]. Dysfunctional gonads can in women with CAH be caused by several factors such as high adrenal androgens and steroid precursors or polycystic ovaries and rarely ovarian rest tumors [76]. In fact, females with NC CAH are sometimes misdiagnosed with polycystic ovary syndrome [9]. In particular, 17OHP and progesterone binds to the progesterone receptor which gives an effect similar to progestin contraceptive pills and affects ovulation as well as the function of endometrium and the cervical mucus [87]. Moreover, high levels of androgens can via aromatization to estrogen suppress the hypothalamic–pituitary–gonadal axis and thus cause hypogonadotropic hypogonadism. Supraphysiological glucocorticoid replacement can also cause hypogonadotropic hypogonadism [76]. However, other factors may play a role in the ability and wish to become pregnant, e.g., virilized genitalia and subsequent genital surgery [88,89] as well as the non-heterosexual orientation [90] which has a direct correlation to the severity of the CYP21A2 genotype [91]. Furthermore, women with CAH may be less sexually active [92]. It should be noted that many females with CAH never try to become pregnant [35], especially those with the SW phenotype [93]. Thus, the proportion of mothers with CAH is low [35,36,93-95]. In a recent epidemiological study 25.4% of Swedish females with CAH had at least one biological child compared to 45.8% of matched controls [36]. Comparing the phenotype groups showed that only 8.1% of females with SW form had a child while 41.8% in SV and 40.8% in NC [36], i.e., reproductive outcomes were only impaired in females with SW CAH, which is in accordance with what has been reported by others [93].
- In males with CAH primary gonadal dysfunction can be caused by TARTs and secondary hypogonadotropic hypogonadism [38,72-74,76]. The latter occur due to increased androgen production from the adrenals downregulating the gonadotropins. Men with CAH are also less sexually active compared to matched controls [96]. Thus, also in males with CAH the fertility rate was markedly reduced [38,73,97-100]. Some required in vitro fertilization to become fathers [38]. However, low fertility may be something of the past. In a Swedish study of 221 males with CAH (all above 15 years of age) and 22,100 matched controls the fertility rate in males with CAH born after the introduction of neonatal screening for CAH was not reduced. For patients born before the screening the likelihood to father a child was halved [98].
FERTILITY
- Early diagnosis with neonatal screening may be beneficial and preserve cognitive functions in children with classic CAH [101,102]. On the contrary, children diagnosed on clinical suspicion and not via neonatal screening, had impaired verbal working memory from a young age [103]. However, chronic treatment with glucocorticoids and/or other aspects of having CAH seem to negatively affect verbal and visuo-spatial working memory over time also in cases that were diagnosed via neonatal screening [104].
- Adrenal crises with salt loss and hypoglycemia add to the negative impact on cognitive performance [105]. In general, intelligence in screened patients seem to be within the normal range of the general population [104,106], but poorer in patients with the null genotype [104].
- Recent data on effects on brain structures in children [102] and adolescents as well as adult patients [107,108] show widespread white matter changes in clinically diagnosed women with CAH in addition to reduced hippocampal and cerebellar volumes [74]. Further, a reduction in total brain volume and altered grey matter was identified in the frontal-parietal cortex in both sexes, but no changes were found in limbic structures in young adult patients diagnosed by neonatal screening [108]. This is opposed by the findings in another cohort (in which half underwent neonatal screening) where children with CAH were found to have smaller amygdala and hippocampal volumes in addition to smaller middle frontal grey matter volumes [102].
- Data on cognition and quality of life (QoL) in NC CAH is very limited, whereas carriers of CAH classic pathogenic variants have been found to be less vulnerable to psychological stress [109].
COGNITION AND BRAIN STRUCTURE
- QoL in both females and males with CAH has shown variable results in different studies probably due to methodological differences in measuring, management and populations studied [2,91,96,110-114]. Sexual satisfaction, which also affects QoL, was in adult females with CAH similar to matched controls but was considerable worse in those with the most severe genotype (null) compared to the other genotype groups [92]. Long-term negative effects of genital surgery on genital/clitoral sensitivity can contribute to lower satisfaction with sexual function [115]. In adult males with CAH sexual satisfaction was in general similar to controls/general population [96,116].
- Women with CAH are more negatively affected by the longterm effects of androgen excess related to periods of poor control and/or late diagnosis. They may develop symptoms such as hirsutism and over time they may develop voice issues with a deeper voice quality especially during puberty [117-119].
- Joint issues appear to be more common in patients with CAH than in the general population [30], a minor part may be explained by more autoimmune rheumatic disease [64], but it is more likely due to a hypermobility type of Ehlers Danlos syndrome [120-122]. The CYP21A2 gene partially overlaps with a gene which affects collagen deposition, tenascin-XB (TNXB). Deletions of CYP21A2 that extend into TNXB cause CAH-X and may affect 14% to 15% of patients with 21OHD [123,124].
MISCELLANEOUS
- Due to its complex nature CAH and its treatment may result in several negative long-term outcomes (Table 1). Knowing the pheno- and genotype may help to prognosticate some outcomes such as fertility/fecundity in women with CAH [35,36]. Neonatal screening for CAH with close collaboration between the screening laboratory and clinicians may improve the long-term outcome [125]. New treatments and improved monitoring during follow-up may further improve the long-term health [14, 126]. Regular monitoring of various risk factors and complications are thus essential to identify problems and institute support and treatment at an early stage.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- This study was funded by the Magnus Bergvall foundation, the Stockholm County Council (ALF-SLL), Swedish Research Council (DNR 2021-02440), Region Stockholm (clinical research appointment DNR RS 2019-1140 to Svetlana Lajic), Karolinska Institutet, and Stiftelsen Frimurare Barnhuset i Stockholm.
- 1. Gidlof S, Falhammar H, Thilen A, von Dobeln U, Ritzen M, Wedell A, et al. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol 2013;1:35–42.ArticlePubMed
- 2. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab 2010;95:5110–21.ArticlePubMedPMC
- 3. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia: current insights in pathophysiology, diagnostics, and management. Endocr Rev 2022;43:91–159.ArticlePubMedPMCPDF
- 4. Kocova M, Concolino P, Falhammar H. Characteristics of In2G variant in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 2022;12:788812.ArticlePubMedPMC
- 5. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med 2020;383:1248–61.ArticlePubMed
- 6. Kocova M, Anastasovska V, Falhammar H. Clinical outcomes and characteristics of P30L mutations in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine 2020;69:262–77.ArticlePubMedPMCPDF
- 7. Espinosa Reyes TM, Collazo Mesa T, Lantigua Cruz PA, Agramonte Machado A, Dominguez Alonso E, Falhammar H. Molecular diagnosis of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. BMC Endocr Disord 2020;20:165.ArticlePubMedPMC
- 8. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2018;103:4043–88.ArticlePubMedPMC
- 9. Nordenstrom A, Falhammar H. Management of endocrine disease: diagnosis and management of the patient with nonclassic CAH due to 21-hydroxylase deficiency. Eur J Endocrinol 2019;180:R127–45.ArticlePubMed
- 10. Falhammar H, Wedell A, Nordenstrom A. Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine 2015;50:306–14.ArticlePubMedPDF
- 11. Zetterstrom RH, Karlsson L, Falhammar H, Lajic S, Nordenstrom A. Update on the Swedish newborn screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Int J Neonatal Screen 2020;6:71.ArticlePubMedPMC
- 12. Nordenstrom A, Ahmed S, Jones J, Coleman M, Price DA, Clayton PE, et al. Female preponderance in congenital adrenal hyperplasia due to CYP21 deficiency in England: implications for neonatal screening. Horm Res 2005;63:22–8.ArticlePubMedPDF
- 13. Falhammar H, Nordenstrom A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine 2015;50:32–50.ArticlePubMed
- 14. Nordenstrom A, Falhammar H, Lajic S. Current and novel treatment strategies in children with congenital adrenal hyperplasia. Horm Res Paediatr 2022 Jan 27 [Epub]. https://doi.org/10.1159/000522260.Article
- 15. Nordenstrom A, Lajic S, Falhammar H. Clinical outcomes in 21-hydroxylase deficiency. Curr Opin Endocrinol Diabetes Obes 2021;28:318–24.ArticlePubMed
- 16. Falhammar H, Thoren M. Clinical outcomes in the management of congenital adrenal hyperplasia. Endocrine 2012;41:355–73.ArticlePubMedPDF
- 17. Whittle E, Falhammar H. Glucocorticoid regimens in the treatment of congenital adrenal hyperplasia: a systematic review and meta-analysis. J Endocr Soc 2019;3:1227–45.ArticlePubMedPMCPDF
- 18. Gomes LG, Mendonca BB, Bachega TA. Long-term cardio-metabolic outcomes in patients with classical congenital adrenal hyperplasia: is the risk real? Curr Opin Endocrinol Diabetes Obes 2020;27:155–61.ArticlePubMed
- 19. Eugster EA, Dimeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr 2001;138:26–32.ArticlePubMed
- 20. Muthusamy K, Elamin MB, Smushkin G, Murad MH, Lampropulos JF, Elamin KB, et al. Clinical review: Adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab 2010;95:4161–72.ArticlePubMed
- 21. Van der Kamp HJ, Otten BJ, Buitenweg N, De Muinck Keizer-Schrama SM, Oostdijk W, Jansen M, et al. Longitudinal analysis of growth and puberty in 21-hydroxylase deficiency patients. Arch Dis Child 2002;87:139–44.ArticlePubMedPMC
- 22. Balsamo A, Cicognani A, Baldazzi L, Barbaro M, Baronio F, Gennari M, et al. CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. J Clin Endocrinol Metab 2003;88:5680–8.ArticlePubMedPDF
- 23. Falhammar H, Filipsson H, Holmdahl G, Janson PO, Nordenskjold A, Hagenfeldt K, et al. Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2007;92:110–6.ArticlePubMedPDF
- 24. Falhammar H, Filipsson H, Holmdahl G, Janson PO, Nordenskjold A, Hagenfeldt K, et al. Increased liver enzymes in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr J 2009;56:601–8.ArticlePubMed
- 25. Falhammar H, Filipsson Nystrom H, Wedell A, Thoren M. Cardiovascular risk, metabolic profile, and body composition in adult males with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol 2011;164:285–93.ArticlePubMed
- 26. Finkielstain GP, Kim MS, Sinaii N, Nishitani M, Van Ryzin C, Hill SC, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2012;97:4429–38.ArticlePubMedPMC
- 27. Falhammar H, Frisen L, Hirschberg AL, Norrby C, Almqvist C, Nordenskjold A, et al. Increased cardiovascular and metabolic morbidity in patients with 21-hydroxylase deficiency: a Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab 2015;100:3520–8.ArticlePubMed
- 28. Torky A, Sinaii N, Jha S, Desai J, El-Maouche D, Mallappa A, et al. Cardiovascular disease risk factors and metabolic morbidity in a longitudinal study of congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021;106:e5247–57.ArticlePubMedPMCPDF
- 29. Tamhane S, Rodriguez-Gutierrez R, Iqbal AM, Prokop LJ, Bancos I, Speiser PW, et al. Cardiovascular and metabolic outcomes in congenital adrenal hyperplasia: a systematic review and meta-analysis. J Clin Endocrinol Metab 2018;103:4097–103.ArticlePubMed
- 30. Falhammar H, Claahsen-van der Grinten H, Reisch N, Slowikowska-Hilczer J, Nordenstrom A, Roehle R, et al. Health status in 1040 adults with disorders of sex development (DSD): a European multicenter study. Endocr Connect 2018;7:466–78.ArticlePubMedPMC
- 31. Bonfig W, Roehl FW, Riedl S, Dorr HG, Bettendorf M, Bramswig J, et al. Blood pressure in a large cohort of children and adolescents with classic adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Am J Hypertens 2016;29:266–72.ArticlePubMed
- 32. Lim SG, Lee YA, Jang HN, Kong SH, Ahn CH, Kim SW, et al. Long-term health outcomes of Korean adults with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 2021;12:761258.ArticlePubMedPMC
- 33. Kim JH, Choi S, Lee YA, Lee J, Kim SG. Epidemiology and long-term adverse outcomes in Korean patients with congenital adrenal hyperplasia: a nationwide study. Endocrinol Metab (Seoul) 2022;37:138–47.ArticlePubMedPMCPDF
- 34. Bachelot A, Golmard JL, Dulon J, Dahmoune N, Leban M, Bouvattier C, et al. Determining clinical and biological indicators for health outcomes in adult patients with childhood onset of congenital adrenal hyperplasia. Eur J Endocrinol 2015;173:175–84.ArticlePubMed
- 35. Hagenfeldt K, Janson PO, Holmdahl G, Falhammar H, Filipsson H, Frisen L, et al. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod 2008;23:1607–13.ArticlePubMed
- 36. Hirschberg AL, Gidlof S, Falhammar H, Frisen L, Almqvist C, Nordenskjold A, et al. Reproductive and perinatal outcomes in women with congenital adrenal hyperplasia: a population-based cohort study. J Clin Endocrinol Metab 2021;106:e957–65.ArticlePubMedPDF
- 37. Badeghiesh A, Ismail S, Baghlaf H, Suarthana E, Dahan MH. Pregnancy, delivery and neonatal outcomes among women with congenital adrenal hyperplasia: a study of a large US database. Reprod Biomed Online 2020;41:1093–9.ArticlePubMed
- 38. Bouvattier C, Esterle L, Renoult-Pierre P, de la Perriere AB, Illouz F, Kerlan V, et al. Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21-hydroxylase deficiency: a French National Survey. J Clin Endocrinol Metab 2015;100:2303–13.ArticlePubMed
- 39. Mooij CF, Kroese JM, Sweep FC, Hermus AR, Tack CJ. Adult patients with congenital adrenal hyperplasia have elevated blood pressure but otherwise a normal cardiovascular risk profile. PLoS One 2011;6:e24204.ArticlePubMedPMC
- 40. Kannel WB, Kannel C, Paffenbarger RS Jr, Cupples LA. Heart rate and cardiovascular mortality: the Framingham Study. Am Heart J 1987;113:1489–94.ArticlePubMed
- 41. Mensink GB, Hoffmeister H. The relationship between resting heart rate and all-cause, cardiovascular and cancer mortality. Eur Heart J 1997;18:1404–10.ArticlePubMed
- 42. Naqvi TZ, Lee MS. Carotid intima-media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging 2014;7:1025–38.ArticlePubMed
- 43. Sartorato P, Zulian E, Benedini S, Mariniello B, Schiavi F, Bilora F, et al. Cardiovascular risk factors and ultrasound evaluation of intima-media thickness at common carotids, carotid bulbs, and femoral and abdominal aorta arteries in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2007;92:1015–8.ArticlePubMed
- 44. Wasniewska M, Balsamo A, Valenzise M, Manganaro A, Faggioli G, Bombaci S, et al. Increased large artery intima media thickness in adolescents with either classical or nonclassical congenital adrenal hyperplasia. J Endocrinol Invest 2013;36:12–5.ArticlePubMed
- 45. Zimmermann A, Sido PG, Schulze E, Al Khzouz C, Lazea C, Coldea C, et al. Bone mineral density and bone turnover in Romanian children and young adults with classical 21-hydroxylase deficiency are influenced by glucocorticoid replacement therapy. Clin Endocrinol (Oxf) 2009;71:477–84.ArticlePubMed
- 46. Sciannamblo M, Russo G, Cuccato D, Chiumello G, Mora S. Reduced bone mineral density and increased bone metabolism rate in young adult patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab 2006;91:4453–8.ArticlePubMedPDF
- 47. Falhammar H, Filipsson H, Holmdahl G, Janson PO, Nordenskjold A, Hagenfeldt K, et al. Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J Clin Endocrinol Metab 2007;92:4643–9.ArticlePubMed
- 48. Falhammar H, Filipsson Nystrom H, Wedell A, Brismar K, Thoren M. Bone mineral density, bone markers, and fractures in adult males with congenital adrenal hyperplasia. Eur J Endocrinol 2013;168:331–41.ArticlePubMed
- 49. El-Maouche D, Collier S, Prasad M, Reynolds JC, Merke DP. Cortical bone mineral density in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 2015;82:330–7.ArticlePubMedPMC
- 50. Espinosa Reyes TM, Leyva Gonzalez G, Dominguez Alonso E, Falhammar H. Bone mass in young patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res Paediatr 2021;94:1–8.ArticlePDF
- 51. Riehl G, Reisch N, Roehle R, Claahsen van der Grinten H, Falhammar H, Quinkler M. Bone mineral density and fractures in congenital adrenal hyperplasia: findings from the dsd-LIFE study. Clin Endocrinol (Oxf) 2020;92:284–94.ArticlePubMedPDF
- 52. Hagenfeldt K, Martin Ritzen E, Ringertz H, Helleday J, Carlstrom K. Bone mass and body composition of adult women with congenital virilizing 21-hydroxylase deficiency after glucocorticoid treatment since infancy. Eur J Endocrinol 2000;143:667–71.ArticlePubMed
- 53. Lee DH, Kong SH, Jang HN, Ahn CH, Lim SG, Lee YA, et al. Association of androgen excess and bone mineral density in women with classical congenital adrenal hyperplasia with 21-hydroxylase deficiency. Arch Osteoporos 2022;17:45.ArticlePubMedPDF
- 54. Rangaswamaiah S, Gangathimmaiah V, Nordenstrom A, Falhammar H. Bone mineral density in adults with congenital adrenal hyperplasia: a systematic review and metaanalysis. Front Endocrinol (Lausanne) 2020;11:493.ArticlePubMedPMC
- 55. Chakhtoura Z, Bachelot A, Samara-Boustani D, Ruiz JC, Donadille B, Dulon J, et al. Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. Eur J Endocrinol 2008;158:879–87.ArticlePubMed
- 56. Chotiyarnwong P, McCloskey EV. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat Rev Endocrinol 2020;16:437–47.ArticlePubMedPDF
- 57. Li L, Bensing S, Falhammar H. Rate of fracture in patients with glucocorticoid replacement therapy: a systematic review and meta-analysis. Endocrine 2021;74:29–37.ArticlePubMedPDF
- 58. Falhammar H, Frisen L, Hirschberg AL, Nordenskjold A, Almqvist C, Nordenstrom A. Increased prevalence of fractures in congenital adrenal hyperplasia: a Swedish population-based national cohort study. J Clin Endocrinol Metab 2022;107:e475–86.ArticlePubMedPMCPDF
- 59. Falhammar H, Butwicka A, Landen M, Lichtenstein P, Nordenskjold A, Nordenstrom A, et al. Increased psychiatric morbidity in men with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014;99:E554–60.ArticlePubMed
- 60. Engberg H, Butwicka A, Nordenstrom A, Hirschberg AL, Falhammar H, Lichtenstein P, et al. Congenital adrenal hyperplasia and risk for psychiatric disorders in girls and women born between 1915 and 2010: a total population study. Psychoneuroendocrinology 2015;60:195–205.ArticlePubMed
- 61. Jenkins-Jones S, Parviainen L, Porter J, Withe M, Whitaker MJ, Holden SE, et al. Poor compliance and increased mortality, depression and healthcare costs in patients with congenital adrenal hyperplasia. Eur J Endocrinol 2018;178:309–20.ArticlePubMed
- 62. Karlsson L, Barbaro M, Ewing E, Gomez-Cabrero D, Lajic S. Epigenetic alterations associated with early prenatal dexamethasone treatment. J Endocr Soc 2018;3:250–63.ArticlePubMedPMC
- 63. Lao Q, Jardin MD, Jayakrishnan R, Ernst M, Merke DP. Complement component 4 variations may influence psychopathology risk in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Genet 2018;137:955–60.ArticlePubMedPMCPDF
- 64. Falhammar H, Frisen L, Hirschberg AL, Nordenskjold A, Almqvist C, Nordenstrom A. Increased risk of autoimmune disorders in 21-hydroxylase deficiency: a Swedish population-based national cohort study. J Endocr Soc 2019;3:1039–52.ArticlePubMedPMCPDF
- 65. Patrova J, Jarocka I, Wahrenberg H, Falhammar H. Clinical outcomes in adrenal incidentaloma: experience from one center. Endocr Pract 2015;21:870–7.ArticlePubMed
- 66. Falhammar H. Non-functioning adrenal incidentalomas caused by 21-hydroxylase deficiency or carrier status? Endocrine 2014;47:308–14.ArticlePubMedPDF
- 67. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016;175:G1–34.ArticlePubMed
- 68. Calissendorff J, Juhlin CC, Sundin A, Bancos I, Falhammar H. Adrenal myelolipomas. Lancet Diabetes Endocrinol 2021;9:767–75.ArticlePubMedPMC
- 69. Falhammar H, Torpy DJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency presenting as adrenal incidentaloma: a systematic review and meta-analysis. Endocr Pract 2016;22:736–52.ArticlePubMed
- 70. Nermoen I, Falhammar H. Prevalence and characteristics of adrenal tumors and myelolipomas in congenital adrenal hyperplasia: a systematic review and meta-analysis. Endocr Pract 2020;26:1351–65.ArticlePubMed
- 71. Kim TM, Kim JH, Jang HN, Choi MH, Cho JY, Kim SY. Adrenal morphology as an indicator of long-term disease control in adults with classic 21-hydroxylase deficiency. Endocrinol Metab (Seoul) 2022;37:124–37.ArticlePubMedPMCPDF
- 72. Engels M, Span PN, van Herwaarden AE, Sweep FC, Stikkelbroeck NM, Claahsen-van der Grinten HL. Testicular adrenal rest tumors: current insights on prevalence, characteristics, origin, and treatment. Endocr Rev 2019;40:973–87.ArticlePubMedPDF
- 73. Falhammar H, Nystrom HF, Ekstrom U, Granberg S, Wedell A, Thoren M. Fertility, sexuality and testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia. Eur J Endocrinol 2012;166:441–9.ArticlePubMedPMC
- 74. Engels M, Gehrmann K, Falhammar H, Webb EA, Nordenstrom A, Sweep FC, et al. Gonadal function in adult male patients with congenital adrenal hyperplasia. Eur J Endocrinol 2018;178:285–94.ArticlePubMed
- 75. Avila NA, Premkumar A, Merke DP. Testicular adrenal rest tissue in congenital adrenal hyperplasia: comparison of MR imaging and sonographic findings. AJR Am J Roentgenol 1999;172:1003–6.ArticlePubMed
- 76. Claahsen-van der Grinten HL, Stikkelbroeck N, Falhammar H, Reisch N. Management of endocrine disease: gonadal dysfunction in congenital adrenal hyperplasia. Eur J Endocrinol 2021;184:R85–97.ArticlePubMed
- 77. Burman P, Falhammar H, Waldenstrom E, Sundin A, Bitzen U. 11C-Metomidate PET/CT detected multiple ectopic adrenal rest tumors in a woman with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021;106:e675. –9.ArticlePubMedPDF
- 78. MacKay D, Nordenstrom A, Falhammar H. Bilateral adrenalectomy in congenital adrenal hyperplasia: a systematic review and meta-analysis. J Clin Endocrinol Metab 2018;103:1767–78.ArticlePubMed
- 79. Falhammar H, Frisen L, Norrby C, Hirschberg AL, Almqvist C, Nordenskjold A, et al. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014;99:E2715–21.ArticlePubMedPDF
- 80. Dorr HG, Wollmann HA, Hauffa BP, Woelfle J; German Society of Pediatric Endocrinology and Diabetology. Mortality in children with classic congenital adrenal hyperplasia and 21-hydroxylase deficiency (CAH) in Germany. BMC Endocr Disord 2018;18:37.ArticlePubMedPMC
- 81. Burger-Stritt S, Kardonski P, Pulzer A, Meyer G, Quinkler M, Hahner S. Management of adrenal emergencies in educated patients with adrenal insufficiency: a prospective study. Clin Endocrinol (Oxf) 2018;89:22–9.ArticlePubMedPDF
- 82. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crisis. N Engl J Med 2019;381:852–61.ArticlePubMed
- 83. Rushworth RL, Torpy DJ, Stratakis CA, Falhammar H. Adrenal crises in children: perspectives and research directions. Horm Res Paediatr 2018;89:341–51.ArticlePubMedPDF
- 84. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crises in older patients. Lancet Diabetes Endocrinol 2020;8:628–39.ArticlePubMed
- 85. Rushworth RL, Chrisp GL, Bownes S, Torpy DJ, Falhammar H. Adrenal crises in adolescents and young adults. Endocrine 2022;77:1–10.ArticlePubMedPMCPDF
- 86. Volkl TM, Ohl L, Rauh M, Schofl C, Dorr HG. Adrenarche and puberty in children with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res Paediatr 2011;76:400–10.ArticlePubMedPDF
- 87. Labarta E, Martinez-Conejero JA, Alama P, Horcajadas JA, Pellicer A, Simon C, et al. Endometrial receptivity is affected in women with high circulating progesterone levels at the end of the follicular phase: a functional genomics analysis. Hum Reprod 2011;26:1813–25.ArticlePubMed
- 88. Nordenskjold A, Holmdahl G, Frisen L, Falhammar H, Filipsson H, Thoren M, et al. Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2008;93:380–6.ArticlePubMed
- 89. Reisch N. Pregnancy in congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2019;48:619–41.ArticlePubMed
- 90. Daae E, Feragen KB, Waehre A, Nermoen I, Falhammar H. Sexual orientation in individuals with congenital adrenal hyperplasia: a systematic review. Front Behav Neurosci 2020;14:38.ArticlePubMedPMC
- 91. Frisen L, Nordenstrom A, Falhammar H, Filipsson H, Holmdahl G, Janson PO, et al. Gender role behavior, sexuality, and psychosocial adaptation in women with congenital adrenal hyperplasia due to CYP21A2 deficiency. J Clin Endocrinol Metab 2009;94:3432–9.ArticlePubMed
- 92. Nordenstrom A, Frisen L, Falhammar H, Filipsson H, Holmdahl G, Janson PO, et al. Sexual function and surgical outcome in women with congenital adrenal hyperplasia due to CYP21A2 deficiency: clinical perspective and the patients’ perception. J Clin Endocrinol Metab 2010;95:3633–40.ArticlePubMed
- 93. Casteràs A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin Endocrinol (Oxf) 2009;70:833–7.ArticlePubMed
- 94. Jaaskelainen J, Hippelainen M, Kiekara O, Voutilainen R. Child rate, pregnancy outcome and ovarian function in females with classical 21-hydroxylase deficiency. Acta Obstet Gynecol Scand 2000;79:687–92.ArticlePubMed
- 95. Słowikowska-Hilczer J, Hirschberg AL, Claahsen-van der Grinten H, Reisch N, Bouvattier C, Thyen U, et al. Fertility outcome and information on fertility issues in individuals with different forms of disorders of sex development: findings from the dsd-LIFE study. Fertil Steril 2017;108:822–31.ArticlePubMed
- 96. Falhammar H, Nystrom HF, Thoren M. Quality of life, social situation, and sexual satisfaction, in adult males with congenital adrenal hyperplasia. Endocrine 2014;47:299–307.ArticlePubMedPDF
- 97. Reisch N, Flade L, Scherr M, Rottenkolber M, Pedrosa Gil F, Bidlingmaier M, et al. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2009;94:1665–70.ArticlePubMedPDF
- 98. Falhammar H, Frisen L, Norrby C, Almqvist C, Hirschberg AL, Nordenskjold A, et al. Reduced frequency of biological and increased frequency of adopted children in males with 21-hydroxylase deficiency: a Swedish population-based national cohort study. J Clin Endocrinol Metab 2017;102:4191–9.ArticlePubMedPDF
- 99. Cabrera MS, Vogiatzi MG, New MI. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001;86:3070–8.ArticlePubMed
- 100. Jaaskelainen J, Kiekara O, Hippelainen M, Voutilainen R. Pituitary gonadal axis and child rate in males with classical 21-hydroxylase deficiency. J Endocrinol Invest 2000;23:23–7.ArticlePubMedPDF
- 101. Messina V, Karlsson L, Hirvikoski T, Nordenstrom A, Lajic S. Cognitive function of children and adolescents with congenital adrenal hyperplasia: importance of early diagnosis. J Clin Endocrinol Metab 2020;105:e683–91.ArticlePubMedPMCPDF
- 102. Herting MM, Azad A, Kim R, Tyszka JM, Geffner ME, Kim MS. Brain differences in the prefrontal cortex, amygdala, and hippocampus in youth with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2020;105:1098–111.ArticlePubMedPMCPDF
- 103. Browne WV, Hindmarsh PC, Pasterski V, Hughes IA, Acerini CL, Spencer D, et al. Working memory performance is reduced in children with congenital adrenal hyperplasia. Horm Behav 2015;67:83–8.ArticlePubMedPMC
- 104. Karlsson L, Gezelius A, Nordenstrom A, Hirvikoski T, Lajic S. Cognitive impairment in adolescents and adults with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2017;87:651–9.ArticlePubMedPDF
- 105. Hamed SA, Metwalley KA, Farghaly HS. Cognitive function in children with classic congenital adrenal hyperplasia. Eur J Pediatr 2018;177:1633–40.ArticlePubMedPDF
- 106. Berenbaum SA, Bryk KK, Duck SC. Normal intelligence in female and male patients with congenital adrenal hyperplasia. Int J Pediatr Endocrinol 2010;2010:853103.ArticlePubMedPMC
- 107. Webb EA, Elliott L, Carlin D, Wilson M, Hall K, Netherton J, et al. Quantitative brain MRI in congenital adrenal hyperplasia: in vivo assessment of the cognitive and structural impact of steroid hormones. J Clin Endocrinol Metab 2018;103:1330–41.ArticlePubMedPMCPDF
- 108. Van’t Westeinde A, Karlsson L, Thomsen Sandberg M, Nordenstrom A, Padilla N, Lajic S. Altered gray matter structure and white matter microstructure in patients with congenital adrenal hyperplasia: relevance for working memory performance. Cereb Cortex 2020;30:2777–88.ArticlePubMedPDF
- 109. Nordenstrom A, Butwicka A, Linden Hirschberg A, Almqvist C, Nordenskjold A, Falhammar H, et al. Are carriers of CYP21A2 mutations less vulnerable to psychological stress?: a population-based national cohort study. Clin Endocrinol (Oxf) 2017;86:317–24.ArticlePubMedPDF
- 110. Strandqvist A, Falhammar H, Lichtenstein P, Hirschberg AL, Wedell A, Norrby C, et al. Suboptimal psychosocial outcomes in patients with congenital adrenal hyperplasia: epidemiological studies in a nonbiased national cohort in Sweden. J Clin Endocrinol Metab 2014;99:1425–32.ArticlePubMed
- 111. Daae E, Feragen KB, Nermoen I, Falhammar H. Psychological adjustment, quality of life, and self-perceptions of reproductive health in males with congenital adrenal hyperplasia: a systematic review. Endocrine 2018;62:3–13.ArticlePubMedPMCPDF
- 112. Verhees MJ, Engels M, Span PN, Sweep FC, van Herwaarden AE, Falhammar H, et al. Quality of life in men with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 2021;12:626646.ArticlePubMedPMC
- 113. Nermoen I, Husebye ES, Svartberg J, Lovas K. Subjective health status in men and women with congenital adrenal hyperplasia: a population-based survey in Norway. Eur J Endocrinol 2010;163:453–9.ArticlePubMed
- 114. Reisch N, Hahner S, Bleicken B, Flade L, Pedrosa Gil F, Loeffler M, et al. Quality of life is less impaired in adults with congenital adrenal hyperplasia because of 21-hydroxylase deficiency than in patients with primary adrenal insufficiency. Clin Endocrinol (Oxf) 2011;74:166–73.ArticlePubMed
- 115. Almasri J, Zaiem F, Rodriguez-Gutierrez R, Tamhane SU, Iqbal AM, Prokop LJ, et al. Genital reconstructive surgery in females with congenital adrenal hyperplasia: a systematic review and meta-analysis. J Clin Endocrinol Metab 2018;103:4089–96.ArticlePubMedPDF
- 116. Gehrmann K, Engels M, Bennecke E, Bouvattier C, Falhammar H, Kreukels BP, et al. Sexuality in males with congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. J Endocr Soc 2019;3:1445–56.ArticlePubMedPMCPDF
- 117. Nygren U, Sodersten M, Falhammar H, Thoren M, Hagenfeldt K, Nordenskjold A. Voice characteristics in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 2009;70:18–25.ArticlePubMed
- 118. Nygren U, Nystrom HF, Falhammar H, Hagenfeldt K, Nordenskjold A, Sodersten M. Voice problems due to virilization in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 2013;79:859–66.ArticlePubMed
- 119. Nygren U, Sodersten M, Thyen U, Kohler B, Nordenskjold A; dsd-LIFE Group. Voice dissatisfaction in individuals with a disorder of sex development. Clin Endocrinol (Oxf) 2019;91:219–27.ArticlePubMedPDF
- 120. Lao Q, Brookner B, Merke DP. High-throughput screening for CYP21A1P-TNXA/TNXB chimeric genes responsible for ehlers-danlos syndrome in patients with congenital adrenal hyperplasia. J Mol Diagn 2019;21:924–31.ArticlePubMedPMC
- 121. Gao Y, Lu L, Yu B, Mao J, Wang X, Nie M, et al. The prevalence of the chimeric TNXA/TNXB gene and clinical symptoms of ehlers-danlos syndrome with 21-hydroxylase deficiency. J Clin Endocrinol Metab 2020;105:dgaa199.ArticlePubMedPDF
- 122. Marino R, Garrido NP, Ramirez P, Notaristefano G, Moresco A, Touzon MS, et al. Ehlers-Danlos syndrome: molecular and clinical characterization of TNXA/TNXB chimeras in congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021;106:e2789–802.ArticlePubMedPDF
- 123. Concolino P, Falhammar H. CAH-X syndrome: genetic and clinical profile. Mol Diagn Ther 2022;26:293–300.ArticlePubMedPDF
- 124. Miller WL, Merke DP. Tenascin-X, congenital adrenal hyperplasia, and the CAH-X syndrome. Horm Res Paediatr 2018;89:352–61.ArticlePubMedPMCPDF
- 125. Lajic S, Karlsson L, Zetterstrom RH, Falhammar H, Nordenstrom A. The success of a screening program is largely dependent on close collaboration between the laboratory and the clinical follow-up of the patients. Int J Neonatal Screen 2020;6:68.ArticlePubMedPMC
- 126. Prete A, Auchus RJ, Ross RJ. Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur J Endocrinol 2021;186:R1–14.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Increased Prevalence of Accidents and Injuries in Congenital Adrenal Hyperplasia: A Population-based Cohort Study
Henrik Falhammar, Angelica Lindén Hirschberg, Agneta Nordenskjöld, Henrik Larsson, Anna Nordenström
The Journal of Clinical Endocrinology & Metabolism.2024; 109(3): e1175. CrossRef - International Newborn Screening Practices for the Early Detection of Congenital Adrenal Hyperplasia
Tracey A. Conlon, Colin P. Hawkes, Jennifer J. Brady, J. Gerard Loeber, Nuala Murphy
Hormone Research in Paediatrics.2024; 97(2): 113. CrossRef - Low renin forms of monogenic hypertension: review of the evidence
Ugochi Chinenye Okorafor, Uchechi Chioma Okorafor
Journal of Clinical Medicine of Kazakhstan.2024; 21(1): 14. CrossRef - Increased risk of nephrolithiasis: an emerging issue in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
Mariangela Chiarito, Crescenza Lattanzio, Vito D’Ascanio, Donatella Capalbo, Paolo Cavarzere, Anna Grandone, Francesca Aiello, Giorgia Pepe, Malgorzata Wasniewska, Thomas Zoller, Mariacarolina Salerno, Maria Felicia Faienza
Endocrine.2024;[Epub] CrossRef - Congenital adrenal hyperplasia: New biomarkers and adult treatments
Bleuenn Dreves, Yves Reznik, Antoine Tabarin
Annales d'Endocrinologie.2023; 84(4): 472. CrossRef - Interpretation of Steroid Biomarkers in 21-Hydroxylase Deficiency and Their Use in Disease Management
Kyriakie Sarafoglou, Deborah P Merke, Nicole Reisch, Hedi Claahsen-van der Grinten, Henrik Falhammar, Richard J Auchus
The Journal of Clinical Endocrinology & Metabolism.2023; 108(9): 2154. CrossRef - Impact of Newborn Screening on Adult Height in Patients With Congenital Adrenal Hyperplasia (CAH)
Heike Hoyer-Kuhn, Alexander J Eckert, Gerhard Binder, Walter Bonfig, Angelika Dübbers, Stefan Riedl, Joachim Woelfle, Helmuth G Dörr, Reinhard W Holl
The Journal of Clinical Endocrinology & Metabolism.2023; 108(11): e1199. CrossRef - Specialty grand challenge in adrenal endocrinology
Henrik Falhammar
Frontiers in Endocrinology.2023;[Epub] CrossRef - Contexts of care for people with differences of sex development
Alexandra E. Kulle, Martina Jürgensen, Ulla Döhnert, Lisa Malich, Louise Marshall, Olaf Hiort
Medizinische Genetik.2023; 35(3): 181. CrossRef - Cardiovascular risk in Cuban adolescents and young adults with congenital adrenal hyperplasia
Tania M. Espinosa Reyes, Alba Katherine Pesántez Velepucha, Julio Oscar Cabrera Rego, Wendy Valdés Gómez, Emma Domínguez Alonso, Henrik Falhammar
BMC Endocrine Disorders.2023;[Epub] CrossRef - Landscape of Adrenal Tumours in Patients with Congenital Adrenal Hyperplasia
Mara Carsote, Ana-Maria Gheorghe, Claudiu Nistor, Alexandra-Ioana Trandafir, Oana-Claudia Sima, Anca-Pati Cucu, Adrian Ciuche, Eugenia Petrova, Adina Ghemigian
Biomedicines.2023; 11(11): 3081. CrossRef - Editorial: Recent advances in diagnosis and treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency
Semra Çaglar Çetinkaya
Frontiers in Endocrinology.2023;[Epub] CrossRef - Approach of Heterogeneous Spectrum Involving 3beta-Hydroxysteroid Dehydrogenase 2 Deficiency
Andreea Gabriela Nicola, Mara Carsote, Ana-Maria Gheorghe, Eugenia Petrova, Alexandru Dan Popescu, Adela Nicoleta Staicu, Mihaela Jana Țuculină, Cristian Petcu, Ionela Teodora Dascălu, Tiberiu Tircă
Diagnostics.2022; 12(9): 2168. CrossRef - Effetti di Crinecerfont sulla secrezione di ACTH nell’iperplasia surrenalica congenita: uno studio di fase 2
Marianna Rita Stancampiano, Silvia Laura Carla Meroni, Giovanna Weber, Gianni Russo
L'Endocrinologo.2022; 23(6): 662. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite