Long-Term Outcomes of Congenital Adrenal Hyperplasia
Article information

Abstract
A plethora of negative long-term outcomes have been associated with congenital adrenal hyperplasia (CAH). The causes are multiple and involve supra-physiological gluco- and mineralocorticoid replacement, excess adrenal androgens both intrauterine and postnatal, elevated steroid precursor and adrenocorticotropic hormone levels, living with a congenital condition as well as the proximity of the cytochrome P450 family 21 subfamily A member 2 (CYP21A2) gene to other genes. This review aims to discuss the different long-term outcomes of CAH.
INTRODUCTION
Congenital adrenal hyperplasia (CAH) is a rare group of autosomal recessive disorders, the most common being 21-hydroxylase deficiency (21OHD) affecting up to 99% of all CAH cases [1-3]. More than 95% of all cases with 21OHD are due to 10 common pathogenic variants in the cytochrome P450 family 21 subfamily A member 2 (CYP21A2) gene which are due to intergenic recombination events with the CYP21A1P pseudogene [3,4]. The impairment of the 21-hydroxylase enzyme results in variable degree of cortisol and aldosterone deficiency and a concomitant increased production of adrenal androgens [5-7]. CAH can be divided into three phenotypes, i.e., salt-wasting (SW), simple virilizing (SV), and non-classic (NC) CAH, the first two are referred to as classic CAH [8]. There is a good genotype-phenotype correlation [9] and analyzing the steroid precursor 17-hydroxyprogesterone (17OHP) levels is the gold standard to diagnose 21OHD [10]. Neonatal screening for 21OHD is today available in many parts of the world and most cases of classic CAH (incidence 1:10,000–1:20,000) are therefore detected during the first week of life [5,11]. Girls with classic CAH present with varying degrees of virilization of external genitalia. If untreated, both girls and boys with SW CAH may develop life threatening neonatal SW crisis. Males, if not subjected to neonatal screening, are diagnosed due to SW crisis or precocious puberty [3]. Thus, neonatal screening is of particular benefit in males [1,11,12]. However, most cases with NC CAH are missed by neonatal screening [9]. The presenting symptoms of androgen excess in patients with NC CAH are milder than in classic CAH and the diagnosis is usually established later in childhood or in young adults [13].
Prior to the introduction of glucocorticoid replacement in the 1950s survival was not possible in cases with the most severe phenotype [1,8]. After the introduction of glucocorticoid and later mineralocorticoid treatment (mineralocorticoid used if needed) this has been the standard treatment [3,8,14]. The balancing act between symptoms of hyperandrogenism and hypercortisolism is, however, complex. Unfortunately, supra-physiological glucocorticoid doses are typically required for sufficient hypothalamic–pituitary–adrenal axis suppression which will result in unfavorable long-term outcomes [3,8,15,16]. The more long-acting the glucocorticoid is the more it seems to affect different outcomes [17]. However, there are no prospective randomized studies comparing various glucocorticoid regimens to investigate glucocorticoid and androgen effects on long-term outcomes [18]. Moreover, some long-term outcomes are a result of the high androgen exposure during fetal life or due to the proximity of the CYP21A2 gene to other genes.
The aim of this review is to discuss different long-term outcomes in patients with CAH.
GROWTH
Both over- and undertreatment with glucocorticoid results in compromised final height, in males and females with CAH [3]. Meta-analyses have shown that patients lose about 1 standard deviation scores in final height corrected for parental height [19,20]. The achieved final height was somewhat better in more recent studies and mineralocorticoid supplementation and early diagnosis seem to be beneficial [20-22].
CARDIOMETABOLIC DISORDERS
Most studies have found an elevated cardiovascular and metabolic risk in patients with CAH [2,23-33]. In a Swedish registry-based study cardiovascular disease (odds ratio [OR], 2.7), hypertension, hyperlipidemia, atrial fibrillation, venous thromboembolism, obesity, diabetes (mainly type 2) and obstructive sleep disorder were increased in patients with CAH compared to matched controls [27]. Women and patients with the NC phenotype had a generally higher morbidity risk. In a Korean epidemiological study, cardiovascular disease (OR, 1.4), stroke, diabetes, hyperlipidemia and hypertension were more prevalent in patients with CAH compared to matched controls [33]. Venous thromboembolism showed a tendency to be more prevalent as well (P=0.055).
Body mass index (BMI) is often high in patients with CAH [2,23,26,30-32,34]. However, this could partly reflect an increase in lean mass in women with CAH due to high androgen exposure during life [23]. In a meta-analysis of both children and adults with CAH, patients had elevated homeostatic model assessment for insulin resistance levels compared to controls [29]. Others found that gestational diabetes, a prediabetes condition, was more likely in pregnant women with CAH than in matched controls [35-37]; however, when adjusted for, among other things obesity, it was no longer significantly increased [37]. The outcome from studies on blood pressure varies, with some studies showing elevated blood pressure in patients with CAH while others do not confirm this [2,23,25,26,28,31,32,38, 39]. Patients with CAH had higher blood pressure in a meta-analysis including both patients with CAH and matched controls [29]. Especially in children, the risk for hypertension seems elevated, and the relative risk (RR) compared to matched controls appears to decrease with age, especially in females with CAH [27,33]. Others have shown that younger children have less hypertension than adolescents with CAH [10]. However, the highest risk was identified in children below the age of 2 years [28] suggesting that high fludrocortisone doses can cause hypertension in young children while high BMI could be the main cause later in life [10].
If the lipid profile is at a disadvantage in patients with CAH is under debate [2,23,25-28,30,38] and in the previously mentioned meta-analysis similar lipid levels were reported in patients with CAH and matched controls [29].
Elevated heart rate is associated with cardiovascular death, particular in men, independent of other risk factors [40,41]. Just a few more beats per minute (bpm), even within normal range, may increase the risk of cardiovascular events [41]. However, only occasional studies have reported elevated heart rates in subjects with CAH. One study demonstrated higher mean 24-hour heart rate in adults with CAH than in BMI-matched controls (by 3 bpm) [39]. In adult males with CAH only those aged 30 years or older had elevated 24-hour heart rate compared to matched controls (by 13 during the day and 27 bpm during the night, respectively) [25], although the effect may be due to obesity in the patient group.
Carotid intima-media thickening (CIMT) measured with ultrasound, is an easy, non-invasive method to find subclinical atherosclerosis [42]. CIMT has been reported to be increased in both children and adults with CAH compared to controls [29,43,44]. The difference was less evident in children and adolescents than in adults with CAH [29].
BONE HEALTH
Bone mineral density (BMD) has been reported to be anything from increased to decreased but most studies have found low BMD in at least one of the measured locations in both sexes and across all age groups [2,26,45-53]. A recent meta-analysis including 254 patients with CAH and 344 matched controls found a lower BMD in CAH in total body, lumbar spine and femoral neck, respectively [54]. Present or accumulated glucocorticoid dosages were negatively correlated to BMD [50-52,55]. Androgen excess seemed to improve BMD in both adult females and young patients with CAH [50,53]. Furthermore, patients with the classic phenotype had worse BMD compared to those with NC CAH [49]. Even though many studies have evaluated BMD in CAH the association between BMD and fractures is poor, especially in glucocorticoid-induced osteoporosis [56].
Fracture risk in CAH has been less thoroughly studied. In a meta-analysis, including individuals with glucocorticoid replacement therapy, treatment increased the fracture risk [57]. Recent epidemiological studies on the presence of fractures in patients with CAH have been reported [33,58]. A Swedish study identified more fractures in the patient group already at a young age compared to the general population (OR, 1.61; mean age of the CAH group 29.8 years) [58]. However, in a Korean study, more fractures were only found in patients aged 40 years and older (OR, 1.4) [33]. In the Swedish study, only those with classic CAH had an increased fracture rate in contrast to individuals with NC CAH who had a fracture rate similar to matched controls [58]. This is in accordance with the findings of a United States study where patients with NC CAH had a lower fracture rate than patients with classic CAH [49]. However, the increased fracture risk in classic CAH may be something of the past since patients with CAH born after the introduction of neonatal screening for CAH had a similar fracture rate compared to matched controls in contrast to those born before the era of neonatal screening [58]. It could be speculated that an early diagnosis and modern management of CAH are superior in reducing fracture occurrence than prolonged androgen exposure on BMD.
PSYCHIATRIC DISEASES
Only few studies have investigated psychiatric co-morbidity in CAH [30,33,59-61]. The OR of having a psychiatric disorder was found to be between 1.5 and 1.9 compared to controls [33, 59,60], with increased depression [61], alcohol misuse [59,60] and suicidality [30,59]. Reaction to severe stress, and adjustment disorders were more prevalent in females with CAH than in matched controls [60].
AUTOIMMUNE DISORDERS
The CYP21A2 gene is located in a highly immunologically active region; hence, it could be speculated that 21OHD would have an effect on autoimmune disorders. On the other hand, the usage of glucocortiocoid replacement required in most patients with 21OHD [14,16], could be assumed to have an immunomodulating effect [62]. In a study of 145 patients with 21OHD, 3.4% had a concurrent autoimmune disorder [63], while in a pan-European study of disorders of sexual development, 22.2% of 222, mostly female patients with CAH, had a concurrent autoimmune disorder [30].
The frequency of autoimmune disorders was demonstrated to be increased in individuals with 21OHD compared to age- and sex-matched controls and increased with age [64]. In this register-based study of 714 patients with 21OHD and 71,400 controls, 7.4% of patients with 21OHD had at least one autoimmune disorder compared to 5.1% of controls (RR, 1.47). Males with 21OHD had a higher risk than females (RR, 1.64 vs. 1.37), mainly due to the fact that autoimmune disorders are more common in females in general. The most prevalent disease was autoimmune thyroid disease which was increased in both sexes.
TUMORS
In patients with non-functional adrenal incidentalomas, especially bilateral tumors or large adrenal myelolipomas, undiagnosed CAH should be considered [65-68]. In a meta-analysis 0.8% of all adrenal incidentalomas was due to a previously undiagnosed genetically confirmed CAH [69]. In another metaanalysis of patients with CAH a quarter had an adrenal tumor of which a quarter was a myelolipoma [70]. Nearly all of the patients with CAH that had a myelolipoma had a late diagnosis or poorly managed CAH [70]. Fig. 1 illustrates bilateral myeolipomas in a patient with CAH. The 17OHP levels correlate with adrenal tumor size and to avoid over-diagnosis of NC CAH genotyping should be performed [66,69]. Adrenocorticotropic hormone (ACTH) is considered the driver of adrenal hyperplasia and later adrenal tumor development [70,71].
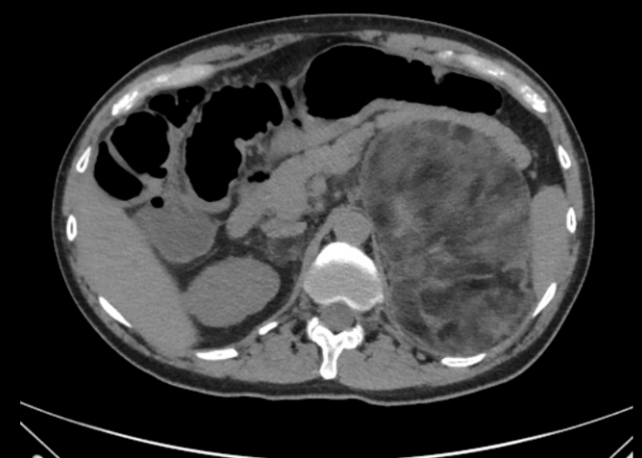
A 18×15×9.5 cm large left-sided and a 2.5×2 cm large right-sided myelolipoma was found in a man in his 40s with simple virilizing congenital adrenal hyperplasia. The hormonal control had been poor for years.
Adrenal rest tumors are common in the testicles of males with CAH and are called testicular adrenal rest tumors (TARTs) [72]. The prevalence of TARTs is around 40% [72], with the highest prevalence reported being 86% [73]. Most TARTs are bilateral [38,73,74]. The prevalence differs depending on the age, severity and probably the ultrasonography equipment if ultrasound of the testicles is performed. Ultrasonography is the preferred method since it is as sensitive as magnetic resonance imaging (MRI) but cheaper and more accessible [75]. Ovarian rest tumors are on the other hand rarely reported [76], possibly due to their location in the abdominal cavity where the use of ultrasonography is not optimal. MRI, or even better positron emission tomography/computer tomography can be used to detect both ovarian and retroperitoneal adrenal rest tumors [77]. ACTH has been proposed to be the most important stimulant of adrenal rest tumors [72]. Thus, intensifying glucocorticoid treatment may reduce the size of adrenal rest tumors in early stages [76] while bilateral adrenalectomy may increase the size [77,78]. It should be noted, though, that there is not always an association between poor control and adrenal rest tumors [32].
MORTALITY
Mortality has not been studied extensively in CAH. In countries without neonatal screening more females than males with SW CAH were diagnosed [12]. However, not only males with SW CAH died undiagnosed in the neonatal period, as was shown by the finding that when neonatal screening was introduced more individuals with SW CAH were found in both sexes [1]. Even when the patients had been diagnosed with CAH the mortality rate was increased with a hazard ratio of 1.6 to 5.17 [33,61,79]. The main cause of death was adrenal crisis, followed by cardiovascular disease (mainly stroke) [79]. However, half of the cases reported as cardiovascular deaths had concomitant severe infection which means that adrenal crisis may be contributing to the majority of the deaths. Moreover, in a survey sent out to German pediatric endocrinologists regarding causes of deaths among their children with CAH, all or almost all deaths were due to adrenal crisis and half had deceased at home [80]. Thus, adrenal crisis can be easily missed both by patients and health professionals inexperienced in adrenal insufficiency [81,82]. Especially in young children and old adults the symptoms can be difficult to interpret [83,84]. However, adolescents and young adults seem to be particularly at risk of adrenal crisis [85].
FERTILITY
Gonadal function has been extensively studied in both sexes in CAH [76]. There are similarities in the gonadal dysfunction between the sexes but also some major discrepancies. In adolescent girls and women with CAH gonadal dysfunction can cause abnormal pubertal development and irregular menses including amenorrhea [76]. The age of pubertal onset and menarche in girls with CAH is similar to controls [35,86]. Since elevated androgen levels and steroid precursors can result in irregular menstruations, the presence of regular menstruations, and especially ovulation, suggest good hormonal control. However, irregular menstruations are also common in the general population and women with CAH may in fact not have a higher frequency [35]. Dysfunctional gonads can in women with CAH be caused by several factors such as high adrenal androgens and steroid precursors or polycystic ovaries and rarely ovarian rest tumors [76]. In fact, females with NC CAH are sometimes misdiagnosed with polycystic ovary syndrome [9]. In particular, 17OHP and progesterone binds to the progesterone receptor which gives an effect similar to progestin contraceptive pills and affects ovulation as well as the function of endometrium and the cervical mucus [87]. Moreover, high levels of androgens can via aromatization to estrogen suppress the hypothalamic–pituitary–gonadal axis and thus cause hypogonadotropic hypogonadism. Supraphysiological glucocorticoid replacement can also cause hypogonadotropic hypogonadism [76]. However, other factors may play a role in the ability and wish to become pregnant, e.g., virilized genitalia and subsequent genital surgery [88,89] as well as the non-heterosexual orientation [90] which has a direct correlation to the severity of the CYP21A2 genotype [91]. Furthermore, women with CAH may be less sexually active [92]. It should be noted that many females with CAH never try to become pregnant [35], especially those with the SW phenotype [93]. Thus, the proportion of mothers with CAH is low [35,36,93-95]. In a recent epidemiological study 25.4% of Swedish females with CAH had at least one biological child compared to 45.8% of matched controls [36]. Comparing the phenotype groups showed that only 8.1% of females with SW form had a child while 41.8% in SV and 40.8% in NC [36], i.e., reproductive outcomes were only impaired in females with SW CAH, which is in accordance with what has been reported by others [93].
In males with CAH primary gonadal dysfunction can be caused by TARTs and secondary hypogonadotropic hypogonadism [38,72-74,76]. The latter occur due to increased androgen production from the adrenals downregulating the gonadotropins. Men with CAH are also less sexually active compared to matched controls [96]. Thus, also in males with CAH the fertility rate was markedly reduced [38,73,97-100]. Some required in vitro fertilization to become fathers [38]. However, low fertility may be something of the past. In a Swedish study of 221 males with CAH (all above 15 years of age) and 22,100 matched controls the fertility rate in males with CAH born after the introduction of neonatal screening for CAH was not reduced. For patients born before the screening the likelihood to father a child was halved [98].
COGNITION AND BRAIN STRUCTURE
Early diagnosis with neonatal screening may be beneficial and preserve cognitive functions in children with classic CAH [101,102]. On the contrary, children diagnosed on clinical suspicion and not via neonatal screening, had impaired verbal working memory from a young age [103]. However, chronic treatment with glucocorticoids and/or other aspects of having CAH seem to negatively affect verbal and visuo-spatial working memory over time also in cases that were diagnosed via neonatal screening [104].
Adrenal crises with salt loss and hypoglycemia add to the negative impact on cognitive performance [105]. In general, intelligence in screened patients seem to be within the normal range of the general population [104,106], but poorer in patients with the null genotype [104].
Recent data on effects on brain structures in children [102] and adolescents as well as adult patients [107,108] show widespread white matter changes in clinically diagnosed women with CAH in addition to reduced hippocampal and cerebellar volumes [74]. Further, a reduction in total brain volume and altered grey matter was identified in the frontal-parietal cortex in both sexes, but no changes were found in limbic structures in young adult patients diagnosed by neonatal screening [108]. This is opposed by the findings in another cohort (in which half underwent neonatal screening) where children with CAH were found to have smaller amygdala and hippocampal volumes in addition to smaller middle frontal grey matter volumes [102].
Data on cognition and quality of life (QoL) in NC CAH is very limited, whereas carriers of CAH classic pathogenic variants have been found to be less vulnerable to psychological stress [109].
MISCELLANEOUS
QoL in both females and males with CAH has shown variable results in different studies probably due to methodological differences in measuring, management and populations studied [2,91,96,110-114]. Sexual satisfaction, which also affects QoL, was in adult females with CAH similar to matched controls but was considerable worse in those with the most severe genotype (null) compared to the other genotype groups [92]. Long-term negative effects of genital surgery on genital/clitoral sensitivity can contribute to lower satisfaction with sexual function [115]. In adult males with CAH sexual satisfaction was in general similar to controls/general population [96,116].
Women with CAH are more negatively affected by the longterm effects of androgen excess related to periods of poor control and/or late diagnosis. They may develop symptoms such as hirsutism and over time they may develop voice issues with a deeper voice quality especially during puberty [117-119].
Joint issues appear to be more common in patients with CAH than in the general population [30], a minor part may be explained by more autoimmune rheumatic disease [64], but it is more likely due to a hypermobility type of Ehlers Danlos syndrome [120-122]. The CYP21A2 gene partially overlaps with a gene which affects collagen deposition, tenascin-XB (TNXB). Deletions of CYP21A2 that extend into TNXB cause CAH-X and may affect 14% to 15% of patients with 21OHD [123,124].
CONCLUSIONS
Due to its complex nature CAH and its treatment may result in several negative long-term outcomes (Table 1). Knowing the pheno- and genotype may help to prognosticate some outcomes such as fertility/fecundity in women with CAH [35,36]. Neonatal screening for CAH with close collaboration between the screening laboratory and clinicians may improve the long-term outcome [125]. New treatments and improved monitoring during follow-up may further improve the long-term health [14, 126]. Regular monitoring of various risk factors and complications are thus essential to identify problems and institute support and treatment at an early stage.

Causes of Short- and Long-Term Outcomes in Patients with Congenital Adrenal Hyperplasia
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was funded by the Magnus Bergvall foundation, the Stockholm County Council (ALF-SLL), Swedish Research Council (DNR 2021-02440), Region Stockholm (clinical research appointment DNR RS 2019-1140 to Svetlana Lajic), Karolinska Institutet, and Stiftelsen Frimurare Barnhuset i Stockholm.