Articles
- Page Path
- HOME > Endocrinol Metab > Volume 38(6); 2023 > Article
-
Brief ReportDiabetes, obesity and metabolism Partial Deletion of Perk Improved High-Fat Diet-Induced Glucose Intolerance in Mice
 Keypoint
Keypoint
In a study using a mouse model, diabetic mice with partial PERK deletion—i.e., Perk+/- mice—showed improved glucose tolerance and insulin secretion when fed a high-fat diet for 23 weeks, without any significant change in obesity or islet size. This improvement correlated with reduced levels of phosphorylated PERK and ATF4 in islets, suggesting a potential therapeutic approach for diet-induced diabetes. -
Jooyeop Lee1*
 , Min Joo Kim1,2*, Seoil Moon1, Ji Yoon Lim1, Kyong Soo Park1, Hye Seung Jung1
, Min Joo Kim1,2*, Seoil Moon1, Ji Yoon Lim1, Kyong Soo Park1, Hye Seung Jung1 -
Endocrinology and Metabolism 2023;38(6):782-787.
DOI: https://doi.org/10.3803/EnM.2023.1738
Published online: November 13, 2023
1Division of Endocrinology and Metabolism, Department of Internal Medicine, Seoul National University Hospital, Seoul, Korea
2Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam, Korea
- Corresponding author: Hye Seung Jung. Department of Internal Medicine, Seoul National University Hospital, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea Tel: +82-2-2072-0240, Fax: +82-2-762-9662, E-mail: jungjhs@gmail.com
- *These authors contributed equally to this work.
Copyright © 2023 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 725 Views
- 45 Download
ABSTRACT
- Although pancreatic endoplasmic reticulum kinase (PERK) is indispensable to beta cells, low-dose PERK inhibitor improved glucose- stimulated insulin secretion (GSIS) and hyperglycemia in diabetic mice. Current study examined if partial deletion of Perk (Perk+/-) recapitulated the effects of PERK inhibitor, on the contrary to the complete deletion. Perk+/- mice and wild-type controls were fed with a high-fat diet (HFD) for 23 weeks. Glucose tolerance was evaluated along with serum insulin levels and islet morphology. Perk+/- mice on normal chow were comparable to wild-type mice in various metabolic features. HFD-induced obesity was not influenced by Perk reduction; however, HFD-induced glucose intolerance was significantly improved since 15-week HFD. HFD-induced compromises in GSIS were relieved by Perk reduction, accompanied by reductions in phosphorylated PERK and activating transcription factor 4 (ATF4) in the islets. Meanwhile, HFD-induced islet expansion was not significantly affected. In summary, partial deletion of Perk improved glucose tolerance and GSIS impaired by diet-induced obesity, without changes in body weights or islet mass.
- Pancreatic endoplasmic reticulum kinase (PERK) is a protein in the endoplasmic reticulum (ER) and plays critical roles in pancreatic beta cells. Genetic compromise of PERK induced pancreatic atrophy and insulin insufficiency, along with early diabetes [1,2].
- In contrast to ablation of Perk expression, chronic treatment of PERK inhibitors GSK2606414 and GSK2656157 at low doses rather improved glucose-stimulated insulin secretion (GSIS) in mouse and human islets [3], improving hyperglycemia in a mouse model of type 2 diabetes [4]. Meanwhile, the PERK inhibitors may have unexpected targets besides PERK: they were proposed to repress receptor-interacting serine/threonine kinase 1 (RIPK1) kinase activity, independently on PERK [5]. Therefore, it is unclear if the anti-diabetic effects of PERK inhibitors were mediated by attenuation of PERK or RIPK1 or both.
- Therefore, we examined if partial reduction of Perk leading to attenuation of PERK activity would recapitulate effects of synthetic PERK inhibitors in mice.
INTRODUCTION
- Animal study
- We used male Perk+/-mice (from Dr. John C. Bell, University of Ottawa) and the wild-type littermates, with mixed background of C57BL/6 and BALB/c. Genotyping by polymerase chain reaction (PCR) demonstrated the wild-type allele and mutant allele as bands of 231 and 302 bp, respectively (primer sets in Supplemental Table S1) [6]. High-fat diet (HFD) was fed for 23 weeks from 5 weeks of age. Insulin levels were measured using an enzyme-linked immunosorbent assay (ELISA) kit (80-INSMS-E01, ALPCO, Salem, NH, USA), from serum obtained before and after intraperitoneal glucose injection (1 g/kg weight). All the animal studies were performed in accordance with the Institutional Animal Care and Use Committee (SNU-190808-1, 22-0281-S1A1).
- Islet expression of unfolded protein response markers
- Mouse islets were isolated as previously, and total RNA or protein were extracted for reverse transcription PCR and Western blotting (Supplemental Tables S2, S3) [4].
- Islet morphology
- After 23-week HFD, pancreas was harvested and embedded in paraffin. Microscopic examination was conducted after hematoxylin and eosin staining and immunohistochemical staining (Supplemental Table S3) using Aperio Digital Pathology and ImageScope (Leica Biosystems, Wetzlar, Germany).
- Statistical analysis
- Data are expressed as mean±standard error of the mean. Statistical analyses were executed using Prism (GraphPad, San Diego, CA, USA). P values <0.05 were considered statistically significant.
METHODS
- Characteristics of Perk+/- mice
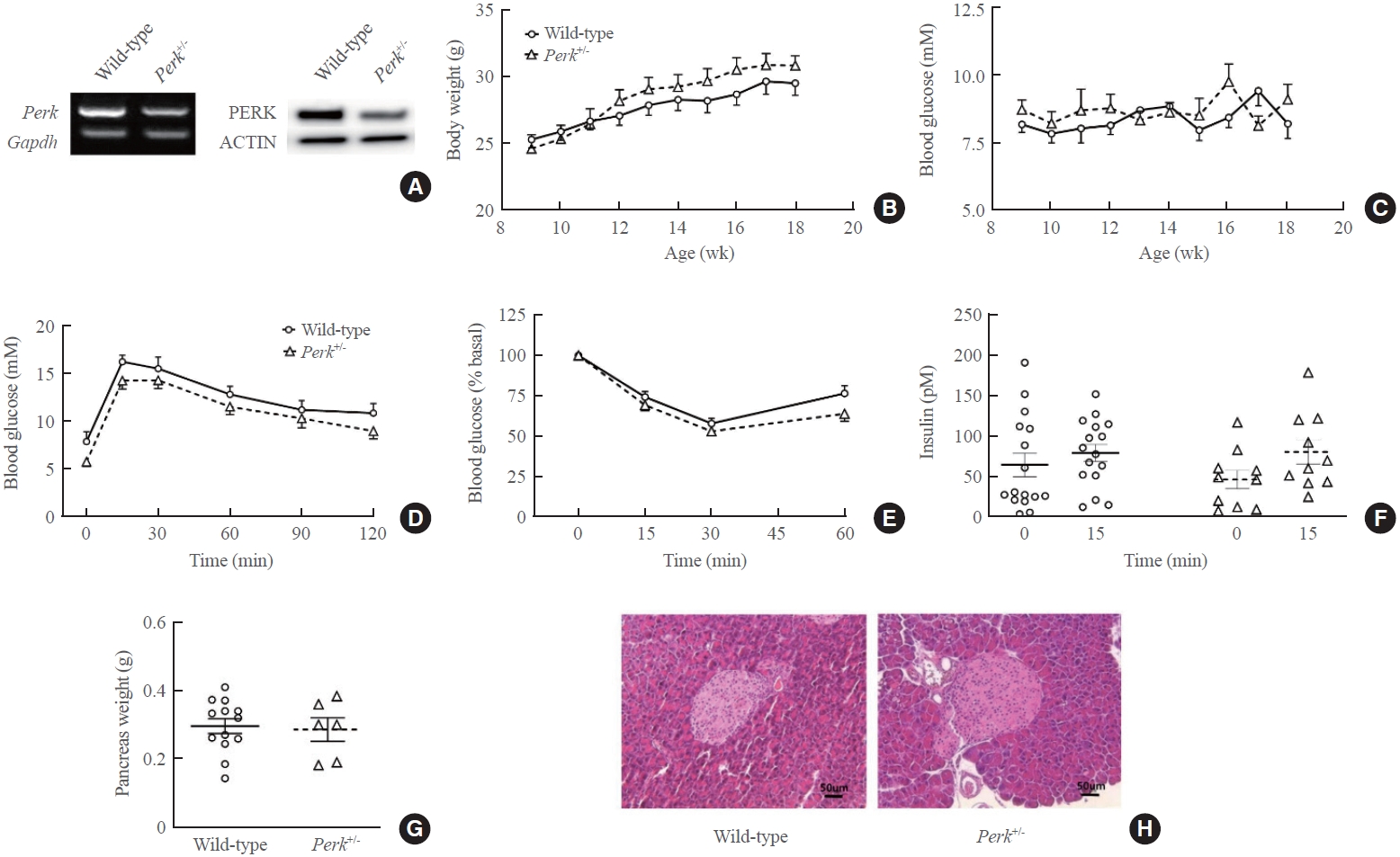
- Reduction of Perk expression was confirmed by RNA and protein levels in islets (Fig. 1A). Body weights, blood glucose levels, glucose tolerance, insulin tolerance, and GSIS were comparable between the genotypes during age of 9 to 18 weeks (Fig. 1B-F). Reduction of Perk did not affect pancreatic weights and islet morphology (Fig. 1G, H).
- Changes in glucose levels by HFD
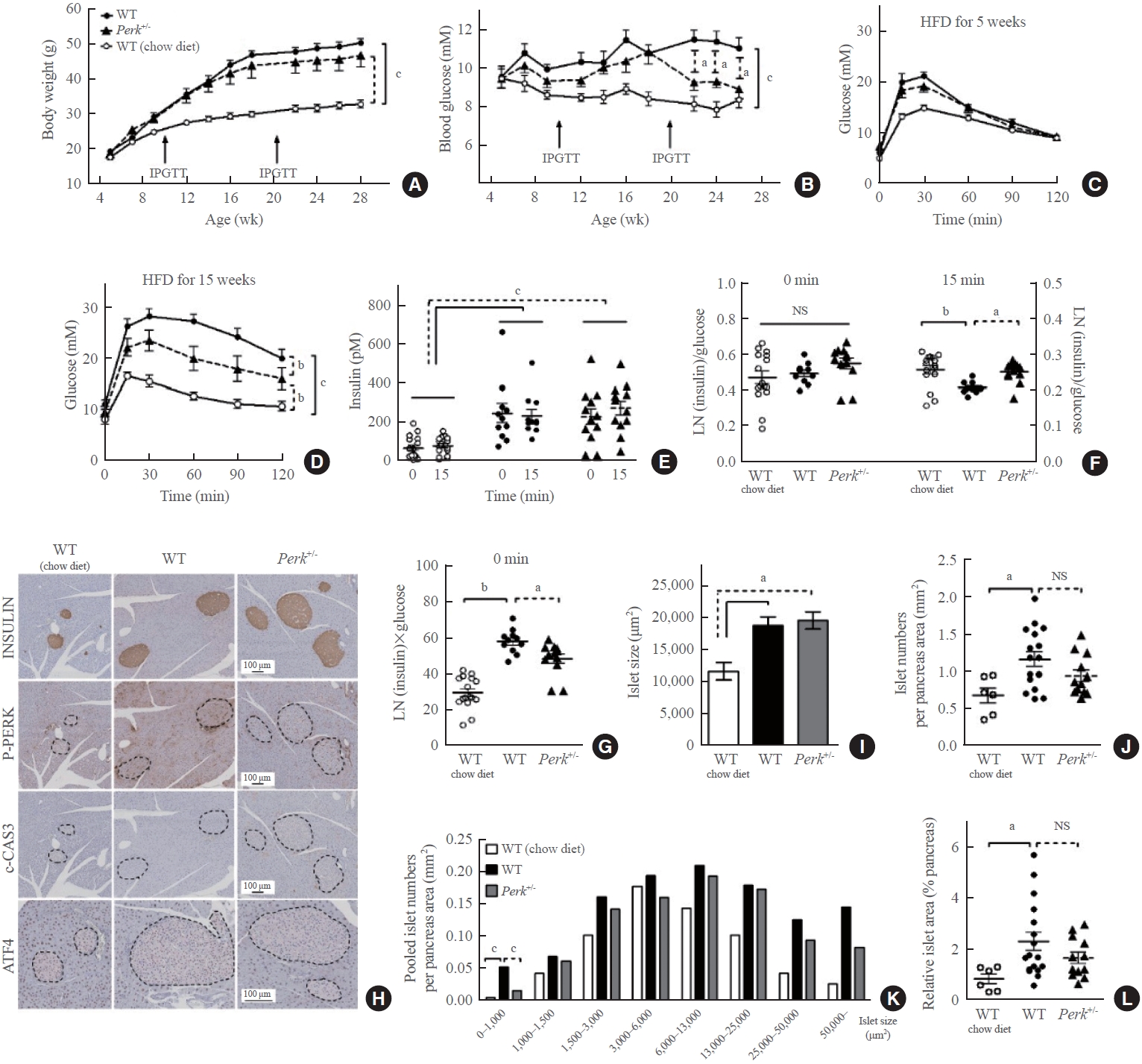
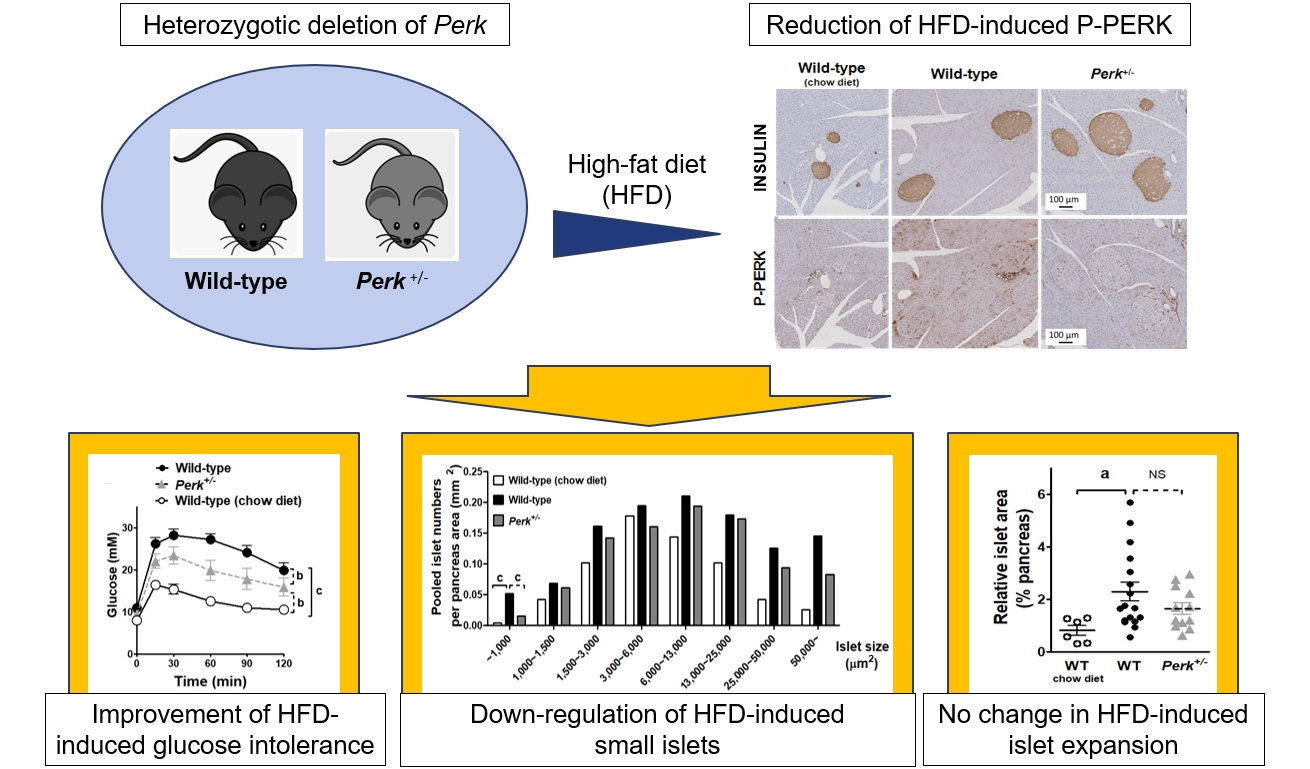
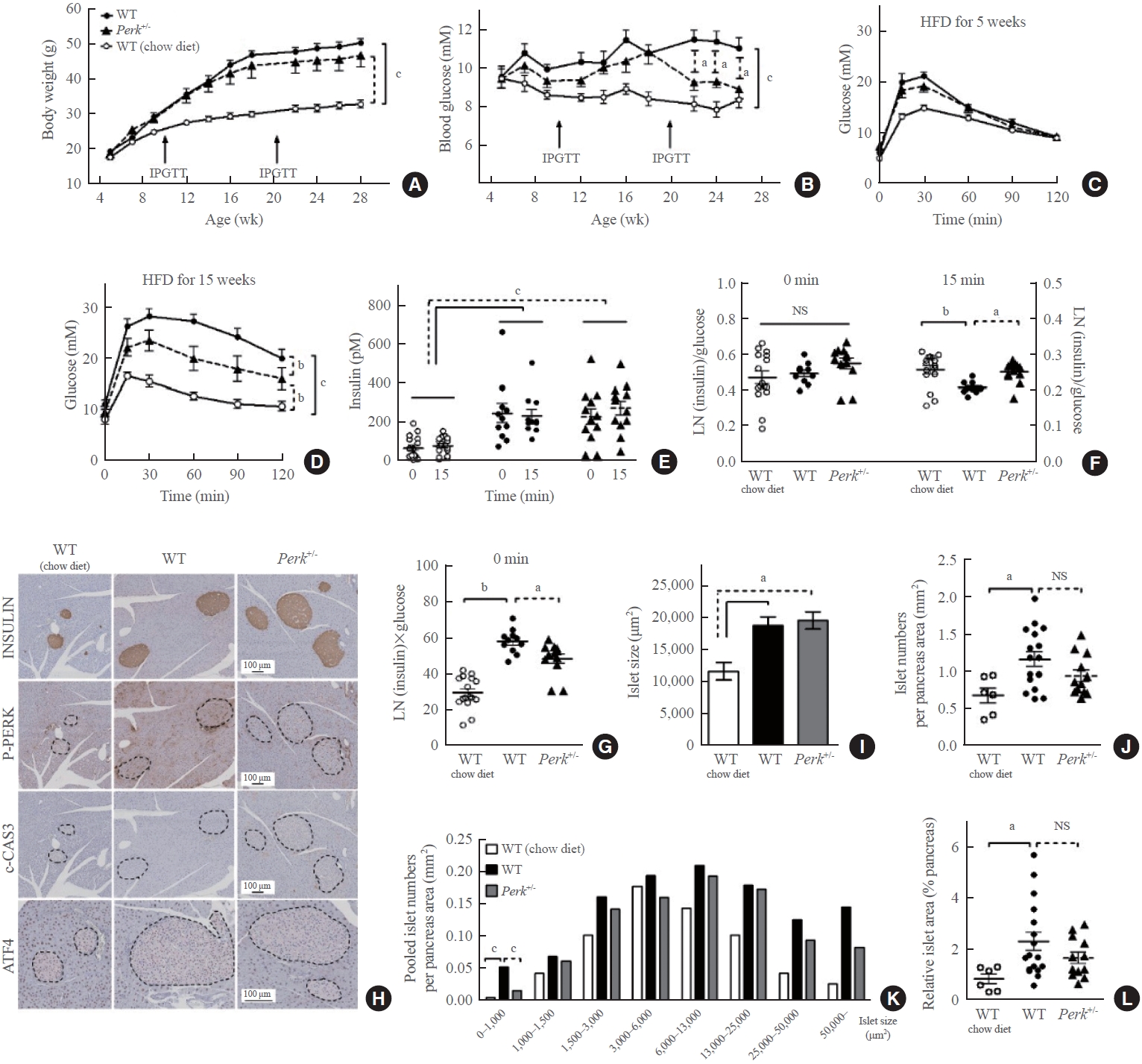
- HFD-induced weight gain without difference between the genotypes (Fig. 2A). HFD-induced hyperglycemia in the wild-type mice (P<0.001) (Fig. 2B). Perk+/- mice showed significantly lower glucose levels compared to the wild-type since 17 weeks of HFD (9.2 mM vs. 11.5 mM in average, P<0.05). HFD-induced glucose intolerance was not affected by Perk reduction after 5-week HFD (Fig. 2C); however, it was significantly improved after 15-week HFD (by 46% of HFD-induced increment in area under the curve, P<0.01) (Fig. 2D).
- Changes in insulin levels by HFD
- The HFD-induced hyperinsulinemia without significant difference between the genotypes (Fig. 2E). Insulin stain intensities on the pancreas sections were also comparable (Fig. 2H). When the plasma insulin levels were adjusted by the corresponding glucose levels, either HFD or Perk reduction did not influence insulin-to-glucose ratio at fasting, while HFD suppressed the ratio after glucose loading, by 20% (P<0.01) (Fig. 2F). It was recovered by Perk reduction (P<0.05). Fasting insulin multiplied by glucose levels suggesting insulin resistance significantly increased by HFD in the control mice by 2-fold, and it was slightly lower in Perk+/- mice than in the wild-type (P<0.05) (Fig. 2G).
- Changes in unfolded protein response markers in the islets by HFD
- According to the immunohistochemical staining, HFD-induced phosphorylated PERK (P-PERK) was significantly down-regulated in the Perk+/- mice. However, cleaved caspase-3 was not induced by HFD in the islets of either genotype (Fig. 2H), indicating that adapted unfolded protein response had prevented ER stress. Nuclear activating transcription factor 4 (ATF4) stain in the islets seemed slightly down-regulated in the Perk+/ mice compared to the wild-type. Reductions in the P-PERK along with phosphorylated eukaryotic translation initiation factor 2A (P-EIF2A) and ATF4, with no significant changes in the final index of ER stress C/EBP homologous protein (CHOP), were reproduced in the Perk+/ islets exposed to chronic lipotoxicity in vitro (Supplemental Fig. S1).
- Changes in islet morphology by HFD
- HFD increased average islet sizes in both the genotypes by 65% (Fig. 2I). HFD increased islet numbers in the wild-type (0.64 vs. 1.11/mm2 pancreas, P<0.05) (Fig. 2J). Despite a tendency to decrease in the numbers by Perk+/- reduction (0.92/mm2), there was no statistical difference between the genotypes. Next, islet numbers according to islet sizes revealed that frequency of small islets (<1,000 μm2) was different from that of the other sizes: more islets in the HFD-fed wild-type, which was significantly inhibited in the Perk+/- mice (P<0.001) (Fig. 2K). Finally, islet area relative to the pancreas area increased in wild-type mice by HFD by 2-fold with an insignificant tendency to decrease by Perk reduction (Fig. 2L).
RESULTS
- In this study, reduction of Perk relieved HFD-induced P-PERK in the islets (Fig. 2H) and impairment of GSIS in mice (Fig. 2F), just like administration of low-dose PERK inhibitor [4]. These findings were accompanied by improvement of glucose intolerance, only after prolonged HFD (Fig. 2B-D), suggesting beta cell exhaustion which could not be reproduced in vitro (Supplemental Fig. S2) [7].
- Initially, Perk+/- mice were reported slightly glucose intolerant [6]. However, Wang et al. [8] found that Perk+/- mice exhibited hyperinsulinemia during neonatal development, causing transient hypoglycemia. They observed that Perk reduction in pancreas, but not in the liver, decreased blood glucose levels. Different genetic backgrounds and ages might explain the conflicting results. In a pathologic environment, Gupta et al. [9] observed that a reduction of Perk dosage in Akita mice with insulin deficient diabetes increased pancreatic insulin contents and improved hyperglycemia, compatible with our current study.
- Expansion of beta cell mass is caused by altered balance between beta cell death and generation [10]. Current study reproduced HFD-induced increases in islet sizes and numbers [11]. Because PERK is critical to beta cells generation [2], the suppression of HFD-induced small islets in Perk+/- mice (Fig. 2K) might reflect inhibition of islet neogenesis due to low PERK activity, and/or secondary to less severe insulin resistance (Fig. 2G). Down-regulation of ATF4 (Fig. 2H, Supplemental Fig. S1) may have a role in the changes in the function and generation of Perk+/- islet. Intriguingly, partial Perk reduction has been reported to increase mature beta cell mass under physiologic condition [8]. PERK inhibitor at low doses did not influence beta cell apoptosis and mass in diabetic mice [4], suggesting that beta cell generation did not change significantly. Further studies are required to answer these complicated questions.
- We also observed that HFD-induced insulin resistance partially improved in the Perk+/- mice (Fig. 2G). P-PERK significantly increased in the liver of obese mice [12], which may decrease insulin sensitivity through forkhead box protein O1 (FOXO1) [13]. Blocking of PERK activity has attenuated fatty acid-induced insulin resistance in hepatocytes [14]. Therefore, the improved insulin resistance might have contributed to the improved glucose intolerance in the Perk+/- mice.
- There are several weak points in this study. Glucose loading did not induce significant insulin levels (Fig. 2E), which reflects inappropriate dose of glucose and/or the measurement time [15]. However, GSIS calculated by insulin-to-glucose ratio could support our hypothesis, instead. Another point is that islet cell composition was not evaluated. Critically, this study cannot tell contribution of RIPK1 in the effects of PERK inhibitors. It requires animal models with RIPK1-insufficient islets. Indeed, inhibition of RIPK1 is known to enhance islet function [16]; therefore, PERK inhibitors might have anti-diabetic effects through attenuation of both PERK and RIPK1.
- In conclusion, attenuation of PERK activity by genetic suppression improved GSIS and hyperglycemia in a mouse model of obesity-induced diabetes, supporting PERK as a new target for diabetes therapy.
DISCUSSION
Supplementary Material
Supplemental Table S1.
Supplemental Table S2.
Supplemental Table S3.
Supplemental Fig. S1.
Supplemental Fig. S2.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: M.J.K., H.S.J. Acquisition, analysis, or interpretation of data: J.L., M.J.K., S.M., J.Y.L. Drafting the work or revising: J.L., K.S.P., H.S.J. Final approval of the manuscript: J.L., M.J.K., S.M., J.Y.L., K.S.P., H.S.J.
Article information
-
Acknowledgements
- This study was supported by NRF (grant number 2022R1A2C 2004570) funded by the Ministry of Science and ICT, Republic of Korea and Seoul National University Hospital Research Fund #0320230310. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
- We are grateful to Drs. John C. Bell (University of Ottawa, Canada) and Seung-Yong Seong (Seoul National University, Korea) for generous provision of Perk+/- mice, and to Dr. Young Joo Park (Seoul National University, Korea) for technical assistance.

- 1. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 2000;25:406–9.ArticlePubMedPDF
- 2. Kefalas G, Larose L. PERK leads a hub dictating pancreatic β cell homoeostasis. Biol Cell 2018;110:27–32.ArticlePubMedPDF
- 3. Kim MJ, Min SH, Shin SY, Kim MN, Lee H, Jang JY, et al. Attenuation of PERK enhances glucose-stimulated insulin secretion in islets. J Endocrinol 2018;236:125–36.ArticlePubMed
- 4. Kim MJ, Kim MN, Min SH, Ham DS, Kim JW, Yoon KH, et al. Specific PERK inhibitors enhanced glucose-stimulated insulin secretion in a mouse model of type 2 diabetes. Metabolism 2019;97:87–91.ArticlePubMed
- 5. Rojas-Rivera D, Delvaeye T, Roelandt R, Nerinckx W, Augustyns K, Vandenabeele P, et al. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ 2017;24:1100–10.ArticlePubMedPMCPDF
- 6. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 2001;7:1153–63.ArticlePubMed
- 7. Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 2005;48:1350–8.ArticlePubMedPDF
- 8. Wang R, Munoz EE, Zhu S, McGrath BC, Cavener DR. Perk gene dosage regulates glucose homeostasis by modulating pancreatic β-cell functions. PLoS One 2014;9:e99684.ArticlePubMedPMC
- 9. Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 2010;59:1937–47.ArticlePubMedPMCPDF
- 10. Sachdeva MM, Stoffers DA. Minireview: meeting the demand for insulin: molecular mechanisms of adaptive postnatal beta-cell mass expansion. Mol Endocrinol 2009;23:747–58.PubMedPMC
- 11. Ahren J, Ahren B, Wierup N. Increased β-cell volume in mice fed a high-fat diet: a dynamic study over 12 months. Islets 2010;2:353–6.ArticlePubMed
- 12. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457–61.ArticlePubMed
- 13. Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev 2013;27:441–9.ArticlePubMedPMC
- 14. Fang Z, Gao W, Jiang Q, Loor JJ, Zhao C, Du X, et al. Targeting IRE1α and PERK in the endoplasmic reticulum stress pathway attenuates fatty acid-induced insulin resistance in bovine hepatocytes. J Dairy Sci 2022;105:6895–908.ArticlePubMed
- 15. Nagy C, Einwallner E. Study of in vivo glucose metabolism in high-fat diet-fed mice using oral glucose tolerance test (OGTT) and insulin tolerance test (ITT). J Vis Exp 2018;131:56672.
- 16. Lau H, Li S, Corrales N, Rodriguez S, Mohammadi M, Alexander M, et al. Necrostatin-1 supplementation to islet tissue culture enhances the in-vitro development and graft function of young porcine islets. Int J Mol Sci 2021;22:8367.ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite