
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 38(3); 2023 > Article
-
Review ArticleCalcium & bone metabolism Skeletal Senescence with Aging and Type 2 Diabetes
 Keypoint
Keypoint
Osteoporosis and type 2 diabetes (T2D) often coexist, leading to poor bone quality and increased fracture risk. Their pathogenesis differs, but emerging evidence indicates shared mechanisms, including senescence, which contributes to many chronic diseases. Cellular senescence impacts various bone-resident cells with age and, as recent research suggests, may also be triggered prematurely in osteocytes by T2D. There is potential in therapeutic interventions that eliminate senescent cells, as they may alleviate age-related bone loss and T2D-induced metabolic dysfunction. Future studies should examine whether these interventions can also mitigate skeletal dysfunction in the context of T2D. -
Joshua Nicholas Farr1,2,3

-
Endocrinology and Metabolism 2023;38(3):295-301.
DOI: https://doi.org/10.3803/EnM.2023.1727
Published online: June 14, 2023
1Robert and Arlene Kogod Center on Aging, Mayo Clinic College of Medicine, Rochester, MN, USA
2Division of Endocrinology, Mayo Clinic College of Medicine, Rochester, MN, USA
3Department of Physiology and Biomedical Engineering, Mayo Clinic College of Medicine, Rochester, MN, USA
- Corresponding author: Joshua Nicholas Farr. Robert and Arlene Kogod Center on Aging, Division of Endocrinology, Department of Physiology and Biomedical Engineering, Mayo Clinic College of Medicine, 200 First Street, SW, Rochester, MN 55905, USA Tel: +1-507-538-0085, Fax: +1-507-284-9111, E-mail: farr.joshua@mayo.edu
• Received: May 5, 2023 • Revised: May 22, 2023 • Accepted: May 26, 2023
Copyright © 2023 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Osteoporosis and type 2 diabetes (T2D) are common diseases that often coexist. While both of these diseases are associated with poor bone quality and increased fracture risk, their pathogenesis of increased fracture risk differs and is multifactorial. Mounting evidence now indicates that key fundamental mechanisms that are central to both aging and energy metabolism exist. Importantly, these mechanisms represent potentially modifiable therapeutic targets for interventions that could prevent or alleviate multiple complications of osteoporosis and T2D, including poor bone quality. One such mechanism that has gained increasing momentum is senescence, which is a cell fate that contributes to multiple chronic diseases. Accumulating evidence has established that numerous boneresident cell types become susceptible to cellular senescence with old age. Recent work also demonstrates that T2D causes the premature accumulation of senescent osteocytes during young adulthood, at least in mice, although it remains to be seen which other bone-resident cell types become senescent with T2D. Given that therapeutically removing senescent cells can alleviate age-related bone loss and T2D-induced metabolic dysfunction, it will be important in future studies to rigorously test whether interventions that eliminate senescent cells can also alleviate skeletal dysfunction in context of T2D, as it does with aging.
- Osteoporosis and type 2 diabetes (T2D) are common diseases that often coexist in the elderly, and in recent years the prevalence and worldwide burden of these diseases has increased dramatically [1,2]. Unfortunately, the available treatments for osteoporosis, T2D, and their complications are suboptimal. Whereas aging is predominantly associated with deficient bone mineral density (BMD) and loss of bone microarchitecture leading to the development of osteoporosis, patients with T2D in contrast tend to have higher BMD yet paradoxically are at increased risk for suffering fragility fractures [3]. Furthermore, the poor bone quality that is characteristic in both osteoporosis and T2D often coincides with the accumulation of several other complications of aging, including cardiovascular disease, cancer, dementia, frailty, infection susceptibility, neuropathy, renal disease, macular degeneration, and sarcopenia [3-5]. The combination of these complications and risk factors significantly increases the development of additional comorbidities and chronic diseases as well as the risk of fractures and death in these individuals [3-5].
- There are numerous pharmacological agents currently available for the treatment of osteoporosis as well as for treating various metabolic bone diseases and skeletal disorders (Table 1). These include the antiresorptive drugs such as hormone replacement therapy (i.e., estrogen and raloxifene), four bisphosphonates (i.e., alendronate, risedronate, ibandronate, and zoledronic acid), and a receptor activator of nuclear factors κB ligand (RANKL) inhibitor (denosumab) as well as two classes of anabolic drugs that include the parathyroid hormone analogues (teriparatide and abaloparatide) and a sclerostin inhibitor (romozosumab) [6]. Although there has been remarkable progress in the treatment of skeletal diseases, these drugs typically only treat bone fragility and not all have been rigorously tested in patients with T2D, whereas aging and T2D are associated with several additional complications (as noted above), thus increasing the prevalence of polypharmacy and the risk of adverse drug interactions in these patients. An alternative approach to the current paradigm is to therapeutically target fundamental hallmark mechanisms that exist at the nexus of osteoporosis and T2D because these diseases are driven by the same underlying pathological mechanisms. By utilizing this strategy, there is now mounting evidence demonstrating that there are key fundamental mechanisms central to energy metabolism and accelerated aging that are potential therapeutic targets for interventions that could slow, prevent, alleviate, or even reverse multiple diseases of aging and complications of T2D [7,8], including poor bone quality and fragility fractures. One such mechanism that has gained increasing momentum in recent years is cellular senescence [9-11].
INTRODUCTION
- In the early 1960’s, work performed by Hayflick and Moorhead [12] established the so-called “Hayflick limit,” which demonstrated that mammalian cells have a finite capacity for cell division resulting in an essentially irreversible permanent cell growth arrest. Since the time of this phenomenal discovery, it has been appreciated by many that diverse forms of age-related stress or metabolic insults can converge to cause a cell to enter this essentially irreversible permanent growth arrest, termed “senescence” [13]. Examples of common stressors that can induce senescence include DNA breaks, reactive oxygen species (ROS), proteotoxic aggregates, and chronic inflammation. The cellular senescence program is activated, for example, by multiple cyclin dependent kinase inhibitors (CDKIs) that can antagonize the actions of CDKs thus resulting in the halt of cell proliferation and in the prevention of malignant transformation [14,15]. Two most notable CDKIs that can be activated to initiate cellular senescence include p16Ink4a (cyclin dependent kinase inhibitor 2A [Cdkn2a]) and p21Cip1 (cyclin dependent kinase inhibitor 1A [Cdkn1a]). Senescent cells consistently develop an altered pattern of gene expression that involves the upregulation of senescent cell anti-apoptotic pathways (SCAPs) [16] as well as a senescence-associated secretory phenotype (SASP) that typically consists of proinflammatory chemokines, cytokines, and matrix degrading/remodeling proteins [17-19]. The SASP can have diverse effects at different stages of life depending on various stimuli and circumstances. Senescent cell accumulation increases in various tissues most commonly with aging, although increased cellular senescence has also been found to occur earlier in life in the contexts of obesity and T2D [9-11]. Under these circumstances, senescent cells presumably accumulate due to metabolic dysfunction, inefficient removal or clearance by the immune system, and because of their resistance to apoptosis or as a result of a combination these factors [20,21]. The biological significance and consequences of senescent cells and their detrimental SASP are becoming more clear as in several models of aging and disease, many laboratories have shown that genetic clearance or pharmacologic killing of senescent cells can improve healthspan (i.e., the period of life free of chronic diseases) and extend lifespan [9-11].
CELLULAR SENESCENCE
- Given that senescent cells accumulate in essentially a universal fashion in several tissues with aging, it is perhaps not surprising that numerous cell types in the bone microenvironment become susceptible to cellular senescence under various scenarios, most notably in response to stress, damaging stimuli, or with old age. Indeed, multiple groups have shown using various approaches and combinations of senescence biomarkers that senescent cells do, in fact, accumulate with aging in old bone tissue and bone marrow where, via their SASP, they are causal in aberrant bone remodeling and in skeletal deterioration. For example, it has been demonstrated that expression of p16Ink4a, which is a key mediator of senescence encoded by the Ink4a/Arf locus [14] (also known as Cdkn2a), increases with aging in various hematopoietic lineage cells (e.g., B-cells and T-cells, myeloid cells) as well as in multiple mesenchymal bone cell lineages (e.g., osteoprogenitors, osteoblasts, and osteocytes [22]) that reside within the bone microenvironment. By contrast, p21Cip1 (also known as Cdkn1a), which is another driver of cellular senescence [15], has been shown to be activated or induced in cells that are under acute stress leading to their senescence in other contexts, such as during wound healing and tissue repair [23,24]. Interestingly, when focusing specifically on the SASP produced by various bone-resident cell populations, it was found that both senescent myeloid lineage cells and senescent osteocytes develop an upregulation of numerous SASP factors with aging [22]. As a whole, the collective data establish that senescent cells are present at the time and location of age-related bone loss and therefore could be causal in the development of osteoporosis.
- In order to establish causality and prove that senescent cells drive age-related bone loss, multiple research laboratories have either utilized transgenic mouse models or have administered drugs (termed “senolytics”) to aged mice to selectively eliminate senescent cells. For example, in one such study [25], senescent cells were removed from old mice using a genetic mouse model called “p16-ATTAC” (i.e., apoptosis through targeted activation of caspase 8), which harbors a “suicide” transgene driven by the p16Ink4a promoter [26]. In addition, a separate cohort of old mice were intermittently treated with senolytics; dasatinib (D; an tyrosine kinase inhibitor used for treating hematologic disorders [27]) plus quercetin (Q; a natural flavanol present in some fruits [28]). Previous studies had shown that the combination of D+Q acts a senolytic cocktail that targets SCAPs to selectively kill senescent cells [29]. After four months of treatment, both the genetic (p16-ATTAC) and pharmacologic (D+Q) approaches were effective in eliminating senescent osteocytes and in preventing the loss of bone quality and strength that occurs with natural chronological aging by reducing bone resorption and improving bone formation [25]. Thus, therapeutically targeting cellular senescence with senolytics may represent an effective strategy to prevent or alleviate age-related bone loss.
CELLULAR SENESCENCE IN BONE
- T2D can cause several features of accelerated aging in both animals and humans. In addition, with obesity and T2D, senescent cells have been shown to accumulate earlier in life in several tissues including, among potentially others, adipose, liver, pancreas, brain, and bone (Fig. 1) where they can prematurely drive several features of aging, at least in mice [30-35]. However, with regards to bone, there are still many unanswered questions regarding which specific cell types are susceptible to cellular senescence in T2D. To address this question, it will first be necessary to develop inducible diabetic animal models that mimic human T2D, specifically in adulthood (after skeletal maturity). Given that T2D often coexists with obesity, these animal models will require high fat diet (HFD) feeding to recapitulate an obesity environment. Furthermore, it will be important for these models to display several hallmark features of human T2D, including hyperglycemia, inadequate insulin secretion, and pancreatic β-cell dysfunction.
- Although several animal models of T2D have been well documented in the literature, one established model that consistently displays these features is the HFD/streptozotocin (STZ) mouse model of T2D, which has been commonly used throughout the diabetes field for many years [36]. With regards to their skeletal phenotype, young adult HFD/STZ (i.e., T2D) mice display several alterations in bone parameters at various skeletal sites that closely mirror those observed in human patients with T2D. For example, one study of HFD/T2D mice found that at 7 months of age, as compared to age-matched non-diabetic controls, that T2D mice have deficient cortical volumetric BMD, thinner cortices, and reduced bone strength as derived by microfinite element analysis [37]. In addition to these impairments in bone microarchitecture, various testing of direct measures of biomechanical strength revealed that bones of T2D mice had reduced ultimate stress and stiffness as examined by spine compression loading or three-point bending of the femur [37]. In addition to bone mass and strength, bone material properties are well recognized as important determinants of bone quality. Interestingly, one test of bone material properties that can be performed on mouse femurs, i.e, cyclic reference point microindentation (RPI) testing, showed that mice with T2D had, on average, higher total indentation distance increase values and lower average loading slope values [37]. These RPI results suggest that, at the tissue-level, bone material properties of T2D mice exhibited reduced fracture toughness and an impaired resistance to microcrack propagation, which both increase the bone’s susceptibility to fracture.
- In order to better understand the underlying cellular changes responsible for the impairments in bone quality, endocortical bone histomorphometry was performed, which revealed that mice with T2D had significantly higher bone resorption (i.e., osteoclast numbers) as compared to age-matched control mice, without a coupled change in the number of osteoblasts [37], which would under normal circumstances be reduced due to coupling between bone resorption and formation. However, despite this maintenance of osteoblasts, bone formation rates were significantly lower on endocortical bone surfaces of T2D mice as compared to age-matched controls, thus demonstrating that the activity of osteoblasts was defective in T2D mice [37]. Therefore, mice treated with HFD/STZ (i.e., T2D mice) have alterations in bone remodeling that lead to deficient bone mass and strength as well as poor bone quality. Importantly, several groups have found similar cortical (using high-resolution peripheral quantitative computed tomography) and bone material property (using in vivo microindentation testing) defects in humans with T2D [38-40].
TYPE 2 DIABETES CAUSES POOR BONE QUALITY AND ALTERS BONE TURNOVER
- Numerous mechanisms have been postulated to be involved in the pathogenesis of skeletal fragility in the context of T2D. Examples of these mechanisms include the damaging effects of prolonged hyperglycemia as well as the accumulation of advanced glycation endproducts (AGEs), proinflammatory factors, oxidative stress, ROS, and cellular senescence [5,11]. In addition, the increased adiposity that is a common feature of T2D has complex effects on bone that can be either beneficial or detrimental in nature [5,11]. For example, while higher body weight and lean soft tissue mass associated with obesity and T2D can have positive mechanical loading effects on weight-bearing skeletal sites, the concomitant increase in circulating adipokines and proinflammatory cytokines, particularly those secreted from visceral adipose tissue stores, can stimulate bone resorption [5,11]. There is also an accumulation of bone marrow adiposity that occurs with obesity and T2D that may have detrimental consequences for the surrounding bone microenvironment, including negative effects on bone formation, although more research is needed to better understand the precise roles of bone marrow adiposity in the pathogenesis of skeletal fragility in T2D.
MECHANISMS OF BONE FRAGILITY WITH TYPE 2 DIABETES
- As noted above, senescent cells have been shown to accumulate in several tissues earlier in life in animal models of T2D [30-35]. These tissues include, but may not limited to, fat, pancreatic β-cells, liver, brain, and bone (Fig. 1) [30-35]. Furthermore, clearance of senescent cells in mice with obesity or T2D has been shown to at least partially alleviate several features of metabolic dysfunction [32,34,41]. Although parallel data in humans with T2D treated with senolytics in still being collected, preliminary studies in patients with diabetic kidney disease treated with senolytics demonstrated efficacy for clearance of senescent cells in adipose tissue and skin as well as reductions in the SASP in blood and fat biopsies [42,43]. From a mechanistic standpoint, senescent cells via their SASP have been shown to contribute to insulin resistance and disrupt insulin signaling by attracting and over-activating immune cells [9]. Examples of immune cell populations that have been implicated include myeloid lineage cells and macrophages that, via their SASP, amplify the accumulation of senescent cells and can spread cellular senescence to neighboring tissues [9]. The SASP produced by non-immune cells can also spread senescence to previously healthy cells [44,45], including to osteocytes in bone [46]. However, whether this also occurs in bone in the context of T2D remains to be tested.
- Notwithstanding, recent studies in mice have already begun to examine the extent to which senescent cells accumulate in bone with T2D, as they do with aging. For example, in young adult mice exposed to HFD/STZ (i.e., T2D mice), osteocyte-enriched bone samples were shown to have elevated mRNA expression of the key mediators of senescence, p16Ink4a and p21Cip1, as compared osteocyte-enriched samples from age-matched control mice [37]. Furthermore, the senescence-associated distension of satellites (SADS) assay [47], revealed that the percentage of senescent osteocytes (defined as ≥4 SADS per osteocyte) was significantly higher in bone cortices of T2D as compared to age-matched control mice [37]. In addition, the telomere-associated foci (TAF) assay, a robust specific marker of cellular senescence that identifies DNA damage sites co-localized at sites of telomeres [48], confirmed that the percentage of TAF+ osteocytes increased significantly in bone cortices of T2D relative to age-matched control mice [37]. Finally, measures of the SASP (i.e., based on an a priori established panel of 36 factors [22]) in the osteocyte-enriched samples of T2D versus control mice, revealed a unique proinflammatory SASP in osteocytes of T2D mice that was comprised predominantly of increased levels of multiple matrix metalloproteinases (MMPs) as well as significantly higher expression of nuclear factor-κB (NF-κB) [37]. It is noteworthy that NF-κB is a key downstream target of the receptor for advanced glycation endproducts (RAGE) pathway that is activated by AGEs [49,50], which represent a hallmark mechanism underlying bone destruction in the pathogenesis of T2D [5,11]. Therefore, elevated MMPs and NFκB constitute at least part of the SASP signature of senescent osteocytes unique to T2D [37]. Taken together, these findings establish that T2D results in the premature accumulation of senescent osteocytes during young adulthood, at least in mice [37], although other bone-resident cell types may also become senescent and additional key SASP factors specific to T2D are likely to be revealed given the rapidly expanding nature of the field. Certainly, future studies are warranted to rigorously test these unanswered questions.
- Since T2D patients have higher fracture risk and poor bone quality [3], and because senescent cells may represent an important mechanistic link between T2D and poor bone quality [11], it will also be important to examine the biological consequences of selectively eliminating senescent cells (using both genetic and pharmacological approaches) on bone microarchitecture, strength, and quality in animal models of T2D. Interestingly, work by Palmer et al. [32] demonstrated in mice that therapeutically targeting senescent cells with senolytics can alleviate obesity-induced metabolic dysfunction. Consistent with these findings, recent studies in mice with T2D by Aguayo-Mazzucato et al. [34] reported that systemic removal of senescent cells, using p16-ATTAC (genetic model of senescent cell elimination) or administration of senolytics (pharmacologic killing of senescent cells) to young adult mice improved insulin secretion and glucose homeostasis. Furthermore, even more recently, Wang et al. [41] reported, using a novel transgenic strategy, that the selective elimination of p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obese mice. However, in each of the above studies, the skeletal phenotype of these animals was not examined. Therefore, it will be important in future studies to rigorously test whether genetic or pharmacological interventions that eliminate senescent cells to thereby alleviate metabolic dysfunction, can also prevent or reverse skeletal dysfunction in context of T2D, as it does with aging [25,30,31].
SKELETAL CELLULAR SENESCENCE IN TYPE 2 DIABETES
- In conclusion, numerous bone-resident cell types become susceptible to cellular senescence with old age. Furthermore, T2D causes the premature accumulation of senescent osteocytes during young adulthood, at least in mice, although it remains to be seen which other bone-resident cell types become senescent with T2D. Given that therapeutically removing senescent cells can alleviate age-related bone loss and T2D-induced metabolic dysfunction, it will be important in future studies to rigorously test whether interventions that eliminate senescent cells can also alleviate skeletal dysfunction in context of T2D, as it does with aging.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- The study was supported by National Institutes of Health grants: R01 DK128552, P01 AG062413, and R21 AG065868.
Fig. 1.Cellular senescence in type 2 diabetes (T2D). Accelerated cellular senescence has been observed in mice with T2D in adipose tissue, pancreatic β-cells, liver, brain, and bone (among potentially other tissues) (created with BioRender.com).
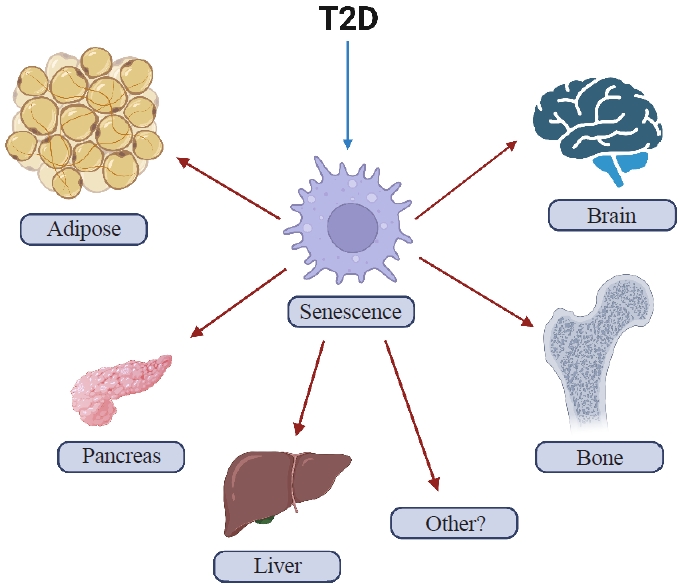
Table 1.Drugs for the Prevention or Treatment of Osteoporosis
- 1. Wright NC, Looker AC, Saag KG, Curtis JR, Delzell ES, Randall S, et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res 2014;29:2520–6.ArticlePubMedPMC
- 2. Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag 2017;20:6–12.ArticlePubMedPMC
- 3. Farr JN, Khosla S. Determinants of bone strength and quality in diabetes mellitus in humans. Bone 2016;82:28–34.ArticlePubMedPMC
- 4. Shanbhogue VV, Mitchell DM, Rosen CJ, Bouxsein ML. Type 2 diabetes and the skeleton: new insights into sweet bones. Lancet Diabetes Endocrinol 2016;4:159–73.ArticlePubMed
- 5. Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol 2017;13:208–19.ArticlePubMedPDF
- 6. Sfeir JG, Drake MT, Khosla S, Farr JN. Skeletal aging. Mayo Clin Proc 2022;97:1194–208.ArticlePubMedPMC
- 7. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194–217.ArticlePubMedPMC
- 8. Farr JN, Almeida M. The spectrum of fundamental basic science discoveries contributing to organismal aging. J Bone Miner Res 2018;33:1568–84.ArticlePubMedPMCPDF
- 9. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol 2020;16:263–75.ArticlePubMedPMCPDF
- 10. Farr JN, Khosla S. Cellular senescence in bone. Bone 2019;121:121–33.ArticlePubMedPMC
- 11. Khosla S, Samakkarnthai P, Monroe DG, Farr JN. Update on the pathogenesis and treatment of skeletal fragility in type 2 diabetes mellitus. Nat Rev Endocrinol 2021;17:685–97.ArticlePubMedPMCPDF
- 12. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585–621.ArticlePubMed
- 13. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966–72.ArticlePubMedPMC
- 14. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 1996;93:13742–7.ArticlePubMedPMC
- 15. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22:4212–22.ArticlePubMedPMC
- 16. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine 2017;21:21–8.ArticlePubMedPMC
- 17. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853–68.PubMed
- 18. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99–118.ArticlePubMedPMC
- 19. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013;15:978–90.ArticlePubMedPMCPDF
- 20. Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol 2018;40:101275.ArticlePubMedPMC
- 21. Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 1995;55:2284–92.PubMed
- 22. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res 2016;31:1920–9.ArticlePubMedPMCPDF
- 23. Saul D, Monroe DG, Rowsey JL, Kosinsky RL, Vos SJ, Doolittle ML, et al. Modulation of fracture healing by the transient accumulation of senescent cells. Elife 2021;10:e69958.ArticlePubMedPMCPDF
- 24. Chandra A, Lagnado AB, Farr JN, Doolittle M, Tchkonia T, Kirkland JL, et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022;21:e13602.ArticlePubMedPMCPDF
- 25. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 2017;23:1072–9.ArticlePubMedPMCPDF
- 26. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011;479:232–6.ArticlePubMedPMCPDF
- 27. Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010;362:2260–70.ArticlePubMed
- 28. D’Andrea G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia 2015;106:256–71.ArticlePubMed
- 29. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015;14:644–58.ArticlePubMedPMCPDF
- 30. Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015;4:e12997.ArticlePubMedPMCPDF
- 31. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24:1246–56.ArticlePubMedPMCPDF
- 32. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019;18:e12950.ArticlePubMedPMCPDF
- 33. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 2017;8:15691.ArticlePubMedPMCPDF
- 34. Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, et al. Acceleration of b cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab 2019;30:129–42.PubMedPMC
- 35. Ogrodnik M, Zhu Y, Langhi LG, Tchkonia T, Kruger P, Fielder E, et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 2019;29:1061–77.ArticlePubMedPMC
- 36. Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, et al. Nongenetic mouse models of non-insulin-dependent diabetes mellitus. Metabolism 1998;47:663–8.ArticlePubMed
- 37. Eckhardt BA, Rowsey JL, Thicke BS, Fraser DG, O’Grady KL, Bondar OP, et al. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 2020;5:e135236.ArticlePubMedPMC
- 38. Farr JN, Drake MT, Amin S, Melton LJ 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J Bone Miner Res 2014;29:787–95.ArticlePubMedPMC
- 39. Furst JR, Bandeira LC, Fan WW, Agarwal S, Nishiyama KK, McMahon DJ, et al. Advanced glycation endproducts and bone material strength in type 2 diabetes. J Clin Endocrinol Metab 2016;101:2502–10.ArticlePubMedPMCPDF
- 40. Nilsson AG, Sundh D, Johansson L, Nilsson M, Mellstrom D, Rudang R, et al. Type 2 diabetes mellitus is associated with better bone microarchitecture but lower bone material strength and poorer physical function in elderly women: a population-based study. J Bone Miner Res 2017;32:1062–71.ArticlePubMedPDF
- 41. Wang L, Wang B, Gasek NS, Zhou Y, Cohn RL, Martin DE, et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab 2022;34:75–89.ArticlePubMedPMC
- 42. Hickson LJ, Langhi Prata LG, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019;47:446–56.ArticlePubMedPMC
- 43. Saul D, Kosinsky RL, Atkinson EJ, Doolittle ML, Zhang X, LeBrasseur NK, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun 2022;13:4827.ArticlePubMedPMCPDF
- 44. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 2012;11:345–9.ArticlePubMedPMCPDF
- 45. Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-kB signalling. Mech Ageing Dev 2018;170:30–6.PubMedPMC
- 46. Farr JN, Saul D, Doolittle ML, Kaur J, Rowsey JL, Vos SJ, et al. Local senolysis in aged mice only partially replicates the benefits of systemic senolysis. J Clin Invest 2023;133:e162519.ArticlePubMedPMC
- 47. Swanson EC, Manning B, Zhang H, Lawrence JB. Higherorder unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol 2013;203:929–42.ArticlePubMedPMCPDF
- 48. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 2012;3:708.ArticlePubMedPMCPDF
- 49. Ramasamy R, Shekhtman A, Schmidt AM. The multiple faces of RAGE: opportunities for therapeutic intervention in aging and chronic disease. Expert Opin Ther Targets 2016;20:431–46.ArticlePubMedPMC
- 50. Litwinoff E, Hurtado Del Pozo C, Ramasamy R, Schmidt AM. Emerging targets for therapeutic development in diabetes and its complications: the RAGE signaling pathway. Clin Pharmacol Ther 2015;98:135–44.ArticlePubMedPMC
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Single-cell sequencing reveals an important role of SPP1 and microglial activation in age-related macular degeneration
Shizhen Lei, Mang Hu, Zhongtao Wei
Frontiers in Cellular Neuroscience.2024;[Epub] CrossRef - The synergistic effect of diabetes mellitus and osteoporosis on the all-cause mortality: a cohort study of an American population
Weihua Li, Siyu Xie, Shengdong Zhong, Liting Lan
Frontiers in Endocrinology.2024;[Epub] CrossRef - Identification of systemic biomarkers and potential drug targets for age-related macular degeneration
Shizhen Lei, Mang Hu, Zhongtao Wei
Frontiers in Aging Neuroscience.2024;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

