Articles
- Page Path
- HOME > Endocrinol Metab > Volume 37(6); 2022 > Article
-
Original ArticleDiabetes, Obesity and Metabolism Gemigliptin Alleviates Succinate-Induced Hepatic Stellate Cell Activation by Ameliorating Mitochondrial Dysfunction
 Keypoint
Keypoint
In this study, gemigliptin decreased the expression of fibrogenesis markers and reduced the abnormal proliferation of hepatic stellate cells. In addition, gemigliptin reduced the succinate-induced production of mitochondrial reactive oxygen species (ROS), intracellular ROS, and mitochondrial fission in hepatic stellate cells (HSCs). Furthermore, in a mouse model of NASH-induced liver fibrosis, gemigliptin alleviated both liver fibrosis and mitochondrial dysfunction. In conclusion, gemigliptin protected against HSC activation and liver fibrosis by alleviating mitochondrial dysfunction and ROS production, indicating its potential as a strategy for preventing the development of liver disease. -
Giang Nguyen*
 , So Young Park*, Dinh Vinh Do, Dae-Hee Choi, Eun-Hee Cho
, So Young Park*, Dinh Vinh Do, Dae-Hee Choi, Eun-Hee Cho -
Endocrinology and Metabolism 2022;37(6):918-928.
DOI: https://doi.org/10.3803/EnM.2022.1530
Published online: November 15, 2022
Department of Internal Medicine, Kangwon National University School of Medicine, Chuncheon, Korea
- Corresponding author: Eun-Hee Cho. Department of Internal Medicine, Kangwon National University School of Medicine, 1 Gangwondaehak-gil, Chuncheon 24341, Korea Tel: +82-33-258-9167, Fax: +82-33-258-2455, E-mail: ehcho@kangwon.ac.kr
- *These authors contributed equally to this work.
Copyright © 2022 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Dipeptidyl peptidase-4 inhibitors (DPP-4Is) are used clinically as oral antidiabetic agents. Although DPP-4Is are known to ameliorate liver fibrosis, the protective mechanism of DPP-4Is in liver fibrosis remains obscure. In this study, gemigliptin was used to investigate the potential of DPP-4Is to alleviate the progression of liver fibrosis.
-
Methods
- To clarify the effects and mechanisms of gemigliptin, we conducted various experiments in LX-2 cells (immortalized human hepatic stellate cells [HSCs], the principal effectors of hepatic fibrogenesis), which were activated by succinate and exhibited elevated expression of α-smooth muscle actin, collagen type 1, and pro-inflammatory cytokines and increased cell proliferation. In vivo, we examined the effects and mechanisms of gemigliptin on a high-fat, high-cholesterol–induced mouse model of nonalcoholic steatohepatitis (NASH).
-
Results
- Gemigliptin decreased the expression of fibrogenesis markers and reduced the abnormal proliferation of HSCs. In addition, gemigliptin reduced the succinate-induced production of mitochondrial reactive oxygen species (ROS), intracellular ROS, and mitochondrial fission in HSCs. Furthermore, in the mouse model of NASH-induced liver fibrosis, gemigliptin alleviated both liver fibrosis and mitochondrial dysfunction.
-
Conclusion
- Gemigliptin protected against HSC activation and liver fibrosis by alleviating mitochondrial dysfunction and ROS production, indicating its potential as a strategy for preventing the development of liver disease.
- Liver fibrosis is a common feature of several chronic liver diseases, such as hepatitis B, hepatitis C, alcoholic hepatitis, autoimmune disorders, and nonalcoholic steatohepatitis (NASH), regardless of their etiology, and is characterized by excessive accumulation of extracellular matrix (ECM) [1,2]. Hepatic stellate cells (HSCs) are the principal collagen-producing cells involved in the production of ECM proteins under conditions of persistent hepatic injury [3]. In the normal liver, HSCs reside in the liver within the space of Disse in a quiescent form, storing vitamin A in lipid droplets. However, following chronic liver damage, HSCs become activated or transdifferentiate into myofibroblast-like cells with proliferative, contractile, migratory, pro-inflammatory, and fibrogenic properties, suggesting that HSC activation is the central mechanism underlying liver fibrogenesis [4,5].
- Succinate, an intermediate of the tricarboxylic acid cycle, has emerged as an important signaling molecule in liver fibrosis [6]. Although succinate is normally produced in mitochondria, accumulated succinate is released into the extracellular space and acts as a paracrine and endocrine effector by binding its receptor, known as G-protein coupled receptor 91 (GPR91), in HSCs. GPR91 is exclusively expressed in HSCs, and once stimulated by succinate it activates the downstream mitogen-activated protein kinase (MEK1/2) by further promoting the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun [7]. Subsequently, the expression of nuclear transcription factors and production of α-smooth muscle actin (α-SMA) stimulates the activation, proliferation, and migration of HSCs. In addition it inhibits apoptosis in HSCs and induces inflammation, leading to liver fibrosis and NASH [8,9].
- Mitochondria are highly dynamic organelles that generate energy and preserve cellular homeostasis through their production of metabolites. Mitochondria are widespread in the cytosol and regulate various cellular physiological functions [10]. Mitochondria continuously alter their morphology to adapt to a new metabolic milieu, responding to metabolic disturbances through fusion and fission cycles [11-13]. Mitochondrial dynamics are engaged in a variety of biological activities, including redox regulation and cell proliferation [14]. Dynamin-related protein 1 (DRP1), a cytosolic GTPase, is a critical regulator of mitochondrial fission. DRP1 is recruited from the cytoplasm to the mitochondrial outer membrane upon activation, where it binds a mitochondrial membrane adaptor, mitochondrial fission factor (MFF), to cause mitochondrial fission [15]. DRP1 activation is regulated by phosphorylation. ERK2 increases the translocation of DRP1 to mitochondria and promotes mitochondrial fission by phosphorylating DRP1 at serine 616 [16].
- Liver fibrosis and chronic liver diseases progress over a long period of time. Therefore, therapies for these conditions should be well tolerated over time, with good targeting to the liver and few undesirable side effects on other organs. However, no antifibrotic agent has yet been approved in clinical practice [17]. Thus, an alternative approach may be to identify an existing clinical compound that shows antifibrotic activity until new drugs become widely available [18,19]. Dipeptidyl peptidase-4 (DPP-4, also known as CD26) is a serine protease that is ubiquitously expressed in many cell types and occurs in soluble form in the circulatory system as an enzyme that breaks down incretins [20-22]. DPP-4 is also highly expressed in the liver and circulation of patients with nonalcoholic fatty liver disease and NASH. Increasing evidence suggests that DPP-4 is highly involved in the development of chronic liver diseases [23,24]. DPP-4 inhibitors (DPP-4Is), which function as glucose-lowering agents by inhibiting DPP-4, have been used for the treatment of type 2 diabetes mellitus. Previous studies have demonstrated that DPP-4Is attenuated hepatic fibrosis by suppressing activated HSCs in rats [25] and prevented NASH-associated liver fibrosis in mice, independent of their antidiabetic effects [26]. Moreover, DPP-4I has been found to attenuate brain mitochondrial dysfunction and decrease brain oxidative stress levels in high-fat diet-fed rats [27].
- In this study, we investigated the correlation between succinate and mitochondrial dysfunction in HSCs and the ability of gemigliptin to attenuate succinate-induced mitochondrial dysfunction and HSC activation.
INTRODUCTION
- Materials
- The reagents and antibodies used in this study were obtained from the indicated suppliers: succinic acid, N-acetyl-L-cysteine (NAC) from Sigma (St. Louis, MO, USA); gemigliptin from LG Chem (Seoul, Korea); primary antibodies: ERK1/2, p-ERK1/2, DRP1, p-DRP1 (Serine 616), MFF, and p-MFF from Cell Signaling Technology (Richmond, CA, USA); p-DRP1 (Serine 616) from Invitrogen (Waltham, MA, USA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from GeneTex (Irvine, CA, USA), GPR91 from Santa Cruz Biotechnology (Dallas, TX, USA), α-SMA from Abcam (Cambridge, England), and collagen 1 from Sigma-Aldrich (St. Louis, MO, USA).
- Cell culture
- Immortalized human HSCs (LX-2 cells) were kindly provided by professor Ja June Jang, Seoul National University. The LX-2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), which included 10% fetal bovine serum and 100 U/mL penicillin/streptomycin.
- Western blot analysis
- Cells and liver tissues were lysed in a radioimmunoprecipitation assay buffer (Atto Corporation, Tokyo, Japan). The protein content in the total cell lysates was determined by using a BCA protein assay kit (Thermo Scientific Pierce, Rockford, IL, USA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to separate equal amounts of total proteins, which were then transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). After transfer, the membrane was incubated in 5% nonfat dry milk in Tris buffered saline containing 0.1% Tween-20 (TBST) for 1 hour at room temperature. After that, the membranes were washed three times with TBST and incubated with primary antibodies overnight at 4°C or for 2 hours at room temperature, according to the manufacturer’s instructions. The membranes were then incubated with appropriate horseradish peroxide-conjugated secondary antibodies at room temperature for 90 minutes. Subsequently, the blots were incubated with WestGlow PICO PLUS chemiluminescent substrate (Biomax, Seoul, Korea) and images of the blots were captured using a ChemiDoc Imaging System (BioRad Laboratories Inc., Hercules, CA, USA). Image Lab software version 6.1.0 (Bio-Rad Laboratories Inc.) was used to calculate the band intensities.
- Cell proliferation assay
- LX-2 cells were seeded in 24-well plates at a density of 0.5×104 cells per well and cultured at 37°C for 24 hours. The medium was replaced with fresh DMEM containing succinic acid (1,600 μmol/L) with or without gemigliptin and incubated for 24 or 48 hours. Viable cell numbers were estimated using cell counting kit-8 (CCK-8) (Dojindo Molecular Technologies Inc., Rockville, MD, USA), with 50μL of the CCK-8 reagent added to each well. The plates were incubated for 1 hour at 37°C, and the absorbance value (optical density) of each well was measured at 450nm according to the manufacturer’s instructions.
- Measurement of reactive oxygen species
- Intracellular reactive oxygen species (ROS) in LX-2 cells treated with succinic acid (1,600 μmol/L) with or without gemigliptin, and 5 mM NAC for 1 hour were analyzed using DCFDA/H2DCFDA (Abcam, Cambridge, UK, ab113851). After treatment for 1 hour with reagents, LX-2 cells were stained with DCFDA (20 μM) for 45 minutes at 37°C in the dark. Images were obtained using a fluorescence microscope (Olympus, Tokyo, Japan).
- Mitochondrial ROS production was examined using the superoxide indicator MitoSOX Red (M36008, Invitrogen). LX-2 cells were plated and treated with succinic acid (1,600 μmol/L) with or without gemigliptin for 1 hour. Next, the cells were washed with phosphate-buffered saline and then incubated with 5 μM MitoSOX reagent and MitoTracker Green (Beyotime, Shanghai, China) for 30 minutes at 37°C in the dark. Images were obtained using a confocal scanning microscope (Olympus).
- Transmission electron microscopy
- LX-2 cells were plated and treated with succinic acid (1,600 μmol/L) with or without gemigliptin for 1 hour. After incubation, the cells were fixed with 2% glutaraldehyde and 2% paraformaldehyde in phosphate buffer (pH 7.4) for 1 hour at 4°C, and then post-fixed in osmium tetroxide for 40 minutes at 4°C. The cells were dehydrated in graded concentrations of ethanol solutions. The cells were treated with a graded propylene oxide series and embedded in Epon. Ultrathin sections (80 nm) were cut from each block and placed on a copper grid. Finally, the samples were stained with uranyl acetate, followed by lead citrate and then observed using a transmission electron microscope (JEOL-2100F, USA, 200 kV) at the Korea Basic Science Institute (Chuncheon, Korea).
- Animal experiments
- The animal study was approved by the Institutional Animal Care and Use Committee of the National Kangwon University (KW-170517-1) and was conducted in accordance with the applicable guidelines. Four-week-old male C57BL/6N mice were purchased from Doo Yeol Biotech (Seoul, Korea). The 4-weekold mice were acclimated to laboratory conditions for 1 week. All mice were housed in the Kangwon National University animal care facility at ambient temperature (22°C±1°C) with a 12/12-hour light/dark cycle and with free access to water and food. The mice were randomly divided into three groups: (1) the control group was fed a control diet (2018S, Envigo, Indianapolis, IN, USA); (2) the NASH model group was fed a high-fat, high-cholesterol (HFHC) diet; and (3) the treatment group was fed an HFHC diet mixed with 400 mg/kg/day of gemigliptin. The HFHC diet contained 38.25 g of 2018S (a fixed formula diet containing a minimum of 18% protein and 5% fat), 1.25 g of cholesterol, 60 g of cocoa butter, and 0.5 g of cholate. The study duration was 8 weeks. Mice were sacrificed at 4 weeks and the end of the study (8 weeks). Centrifugation was used to separate plasma from blood. All tissue samples were kept at –80°C.
- Hematoxylin and eosin stain and Masson’s trichrome stain
- Mouse liver tissues were fixed in 4% paraformaldehyde for histological analysis. The tissues were embedded in paraffin wax after being dehydrated with a graded series of ethanol solutions. Serial frontal sections were cut and stained with hematoxylin and eosin to grade NASH activity, and Masson’s trichrome to stage fibrosis. Photographs were obtained using a microscope.
- Hepatic malondialdehyde determination
- Liver tissue was placed in a microtube containing buffer (20 mM sodium-phosphate buffer at pH approximately 3–3.2+0.5% TritonX-100) and then homogenized in an icebox using a homogenizer. All the samples were normalized by using a bicinchoninic acid protein assay. Malondialdehyde (MDA) formation in the liver was measured using a Lipid Peroxidation (MDA) Assay kit (ab233471, Abcam, Cambridge, UK).
- Quantitative real time-polymerase chain reaction
- Total RNA was extracted with an AccuPrep Universal RNA Extraction Kit (Bioneer Corp., Daejeon, Korea), and then transcribed to cDNA with Maxime RT PreMix Kit (iNtRON Biotechnology Inc., Seongnam, Korea), according to the manufacturer’s instructions. Real-time polymerase chain reaction (PCR) analysis was performed using Power SYBR Green PCR Master Mix (Life Technologies Ltd., Paisley, UK) on QuantStudio 6 Flex Real-Time PCR System (Life Technologies Holdings Pte Ltd., Singapore) with the primer sequences described below. The comparative cycle threshold (ΔΔCt) method was used to evaluate the mRNA expression levels of target genes, with the cycle threshold values of target genes normalized to GAPDH as an internal standard. The primer sequences were as follows: tumor necrosis factor-α (TNFα) forward primer 5´-ATGGCCTCCCTCTCATCAGT -3´ and reverse primer 5´-TTTGGTACGACGTGGGCTAC-3´; interleukin-1β (IL-1β) forward primer 5´-GAGCACCTTCTTTTCCTTCATCTT-3´ and reverse primer 5´-TCACACACCAGCAGGTTATCATC-3´; GAPDH forward primer 5´-ACTCCACTCACGGCAAATTC-3´ and reverse primer 5´-TCTCCATGGTGGTGAAGACA-3´.
- Statistical analyses
- Results are presented as the mean±standard error of the mean of at least three independent replicate experiments. Differences between the treatment groups were evaluated using the Student t test with Microsoft Excel for Microsoft 365 version 2109 build 16.0.14430.20292 (Microsoft Corp., Redmond, WA, USA). Statistical significance was set at P<0.05.
METHODS
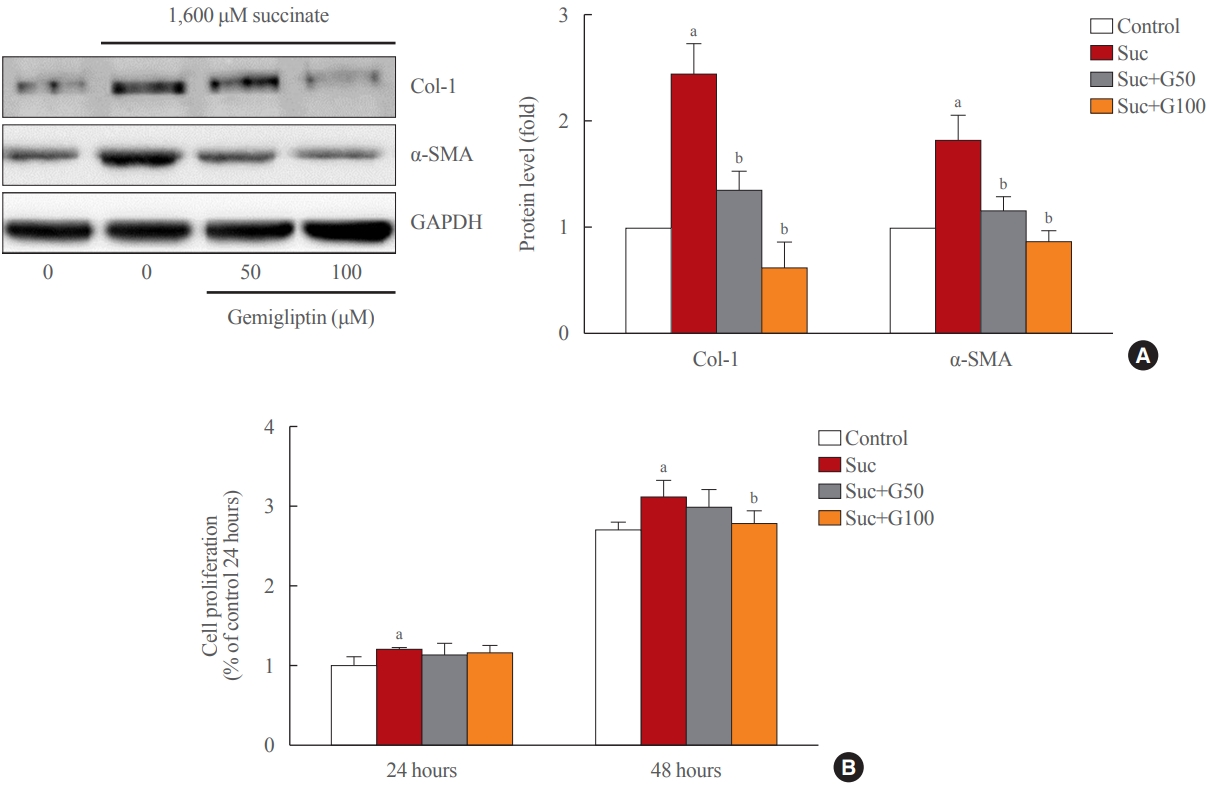
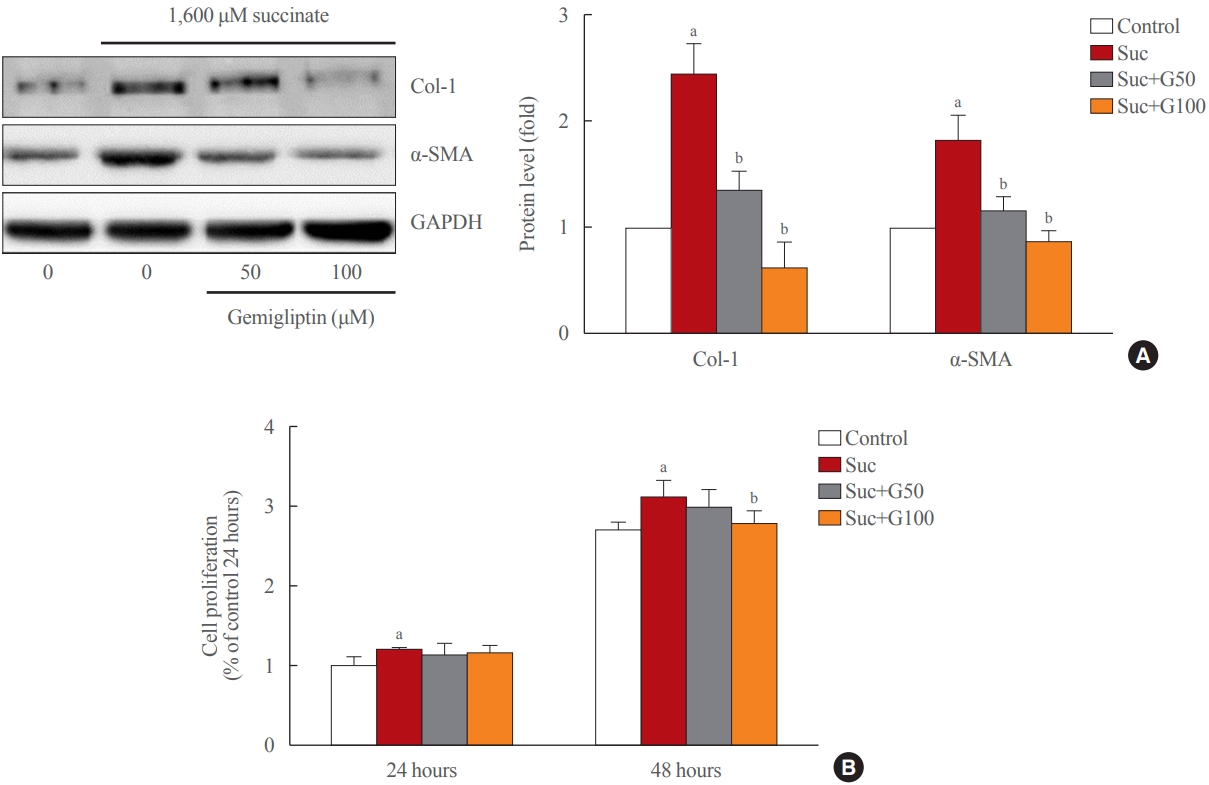
- Gemigliptin ameliorated HSC activation
- As shown in Fig. 1A, treatment of LX-2 cells with succinate for 24 hours caused a significant increase in the expression of fibrogenic proteins (α-SMA, collagen type 1) when compared to that in control cells, and co-treatment with gemigliptin effectively reduced the expression of the fibrogenic proteins in a dose-dependent manner. The proliferation of LX-2 cells at 24 and 48 hours following succinate stimulation increased in a time-dependent manner compared to the control cells (Fig. 1B). However, gemigliptin (100 μM) markedly inhibited succinate-induced HSC proliferation at 48 hours (Fig. 1B). These results suggest that gemigliptin may ameliorate succinate-induced HSC activation.
- Gemigliptin inhibited the succinate-GPR91 signaling pathway in HSCs
- Succinate treatment significantly increased GPR91 expression and p-ERK1/2 signaling pathway in LX-2 cells compared to that in the control (Fig. 2). Interestingly, GPR91 and p-ERK1/2 protein levels were attenuated in LX-2 cells treated with gemigliptin in the presence of succinate (Fig. 2). These data suggest that succinate leads to HSCs activation by directly activating f GPR91 and phosphorylating ERK1/2, and gemigliptin may inhibit HSC activation by inhibiting the succinate-GPR91 pathway.
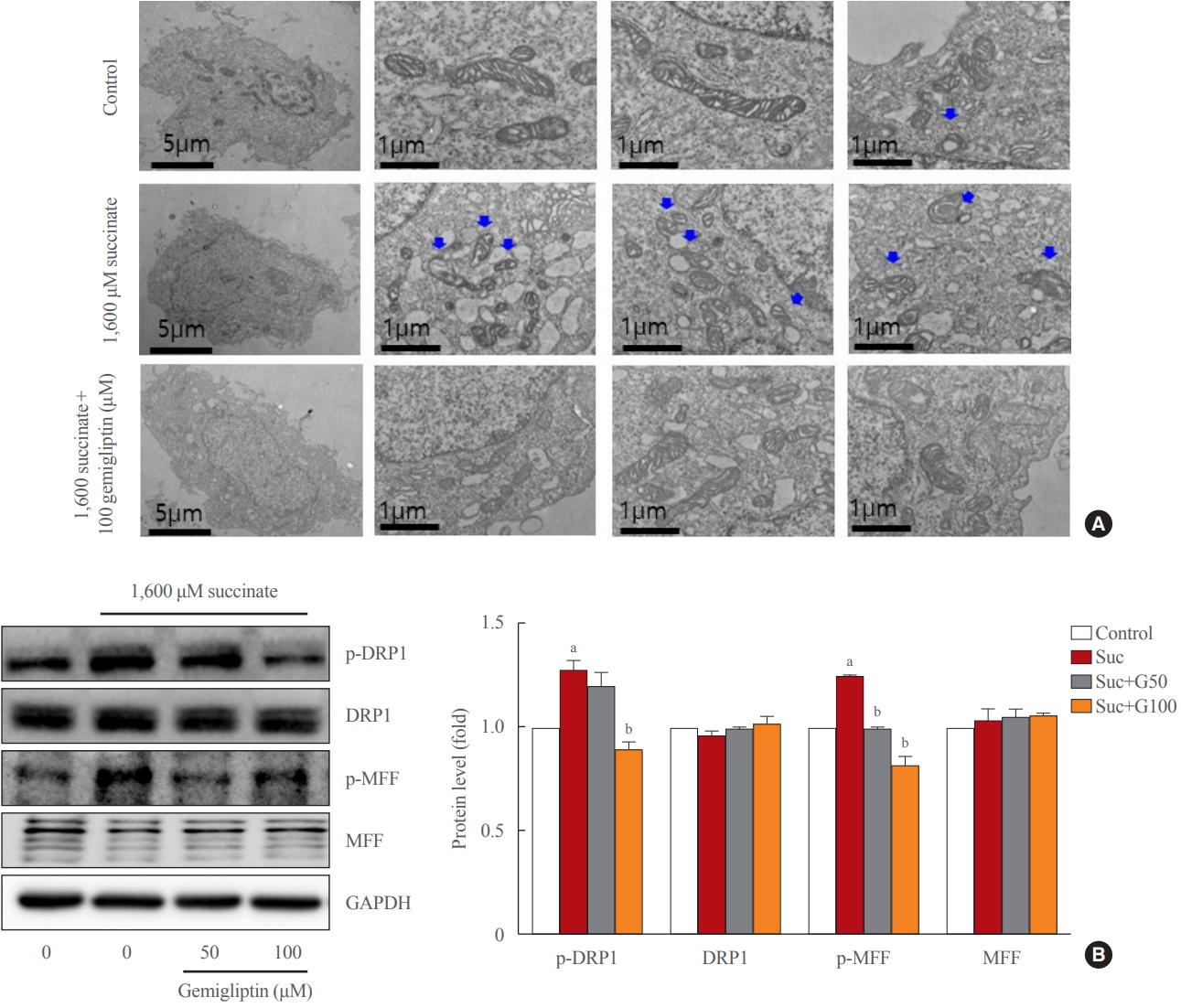
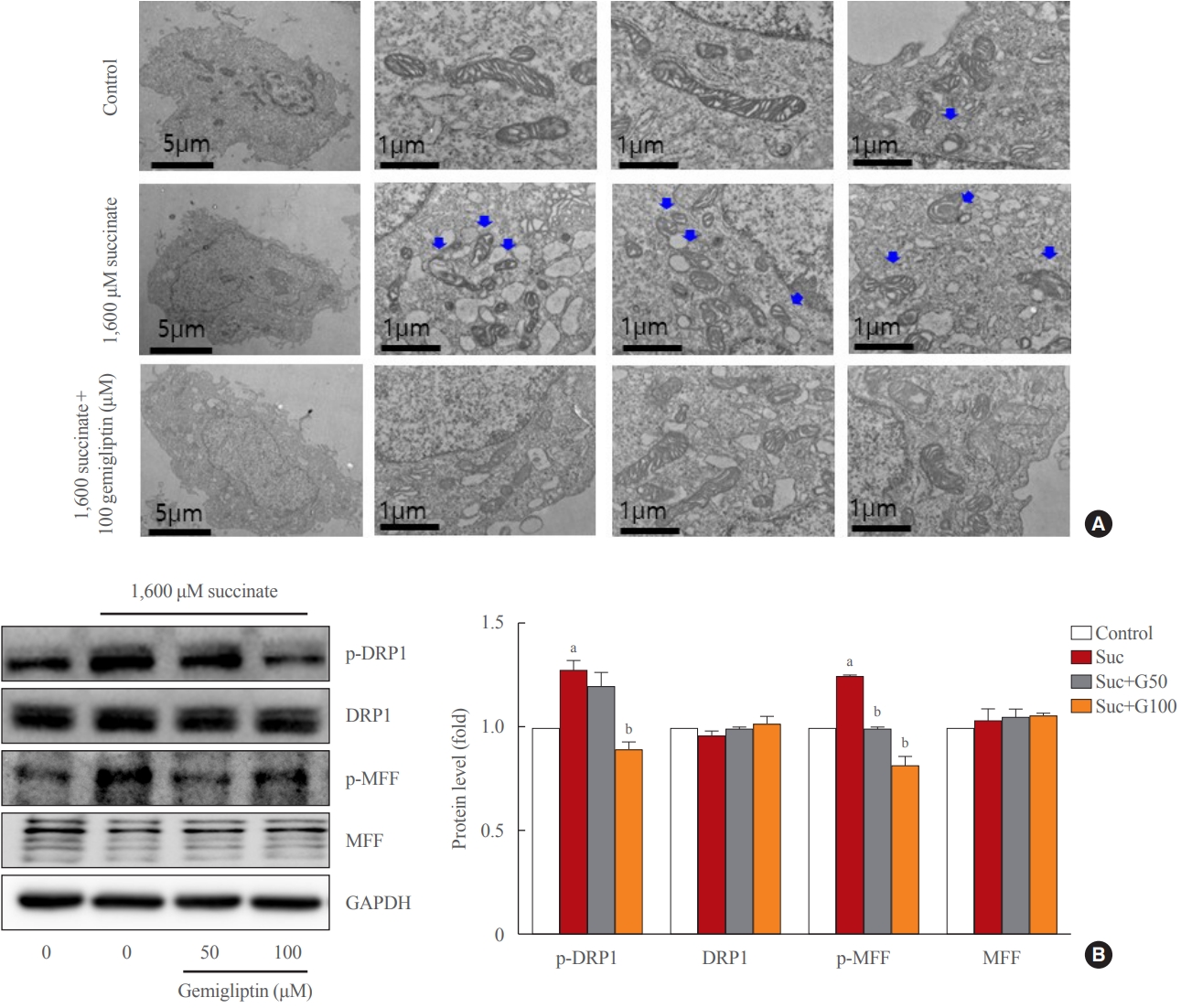
- Gemigliptin alleviated succinate-induced mitochondrial dysfunction
- In a stressful environment, the HSCs were activated; they not only produced ECM proteins, but also generated ROS [28]. Fig. 2B indicated that succinate markedly increased intracellular ROS, and gemigliptin protected HSCs by reducing intracellular ROS in LX-2 cells. Furthermore, we observed that succinate also induced mitochondrial ROS generation, which was attenuated by treatment with gemigliptin for 1 hour (Fig. 2C).
- In normal HSCs, the mitochondria presented long tubular formations, whereas succinate stimulation caused mitochondrial fragmentation, a morphological change referred to as mitochondrial fission (Fig. 3A). Gemigliptin (100 μM) alleviated succinate-induced mitochondrial fission, demonstrating its protective effect on mitochondrial integrity (Fig. 3A). Succinate increased DRP1 phosphorylation (Ser616), a marker of mitochondrial fission, which was reversed by treatment of LX-2 cells with gemigliptin for 1 hour (Fig. 3B). Furthermore, succinate treatment increased the phosphorylation of MFF, which was decreased by gemigliptin treatment (Fig. 3B). These results demonstrate the protective effect of gemigliptin on mitochondrial integrity and ROS production during HSC activation.
- Gemigliptin attenuated liver inflammation and fibrosis
- At 4 weeks of the HFHC diet, significantly increased steatosis was observed in the HFHC diet group, and gemigliptin co-treatment attenuated the steatosis. After 8 weeks of the HFHC diet, there was prominent fibrosis, and the gemigliptin co-treatment group showed less fibrosis than the HFHC diet group (Fig. 4A, B). Western blot results of the 8-week treated mice showed that the HFHC diet enhanced the hepatic expression of collagen type 1 and α-SMA and gemigliptin treatment suppressed α-SMA and collagen type 1 protein levels (Fig. 4C). In addition, the effect of gemigliptin on HFHC-induced hepatic inflammation, as measured by mRNA expression levels of TNFα and IL-1β, was attenuated after 8 weeks (Fig. 4D).
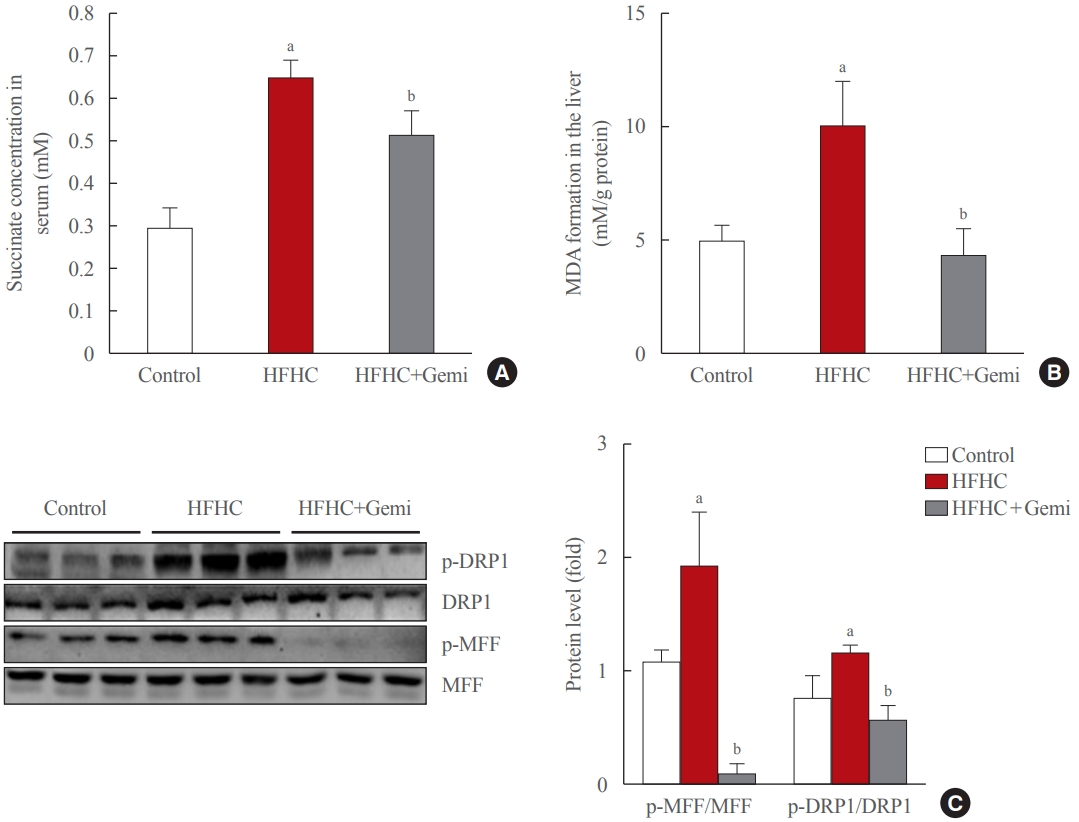
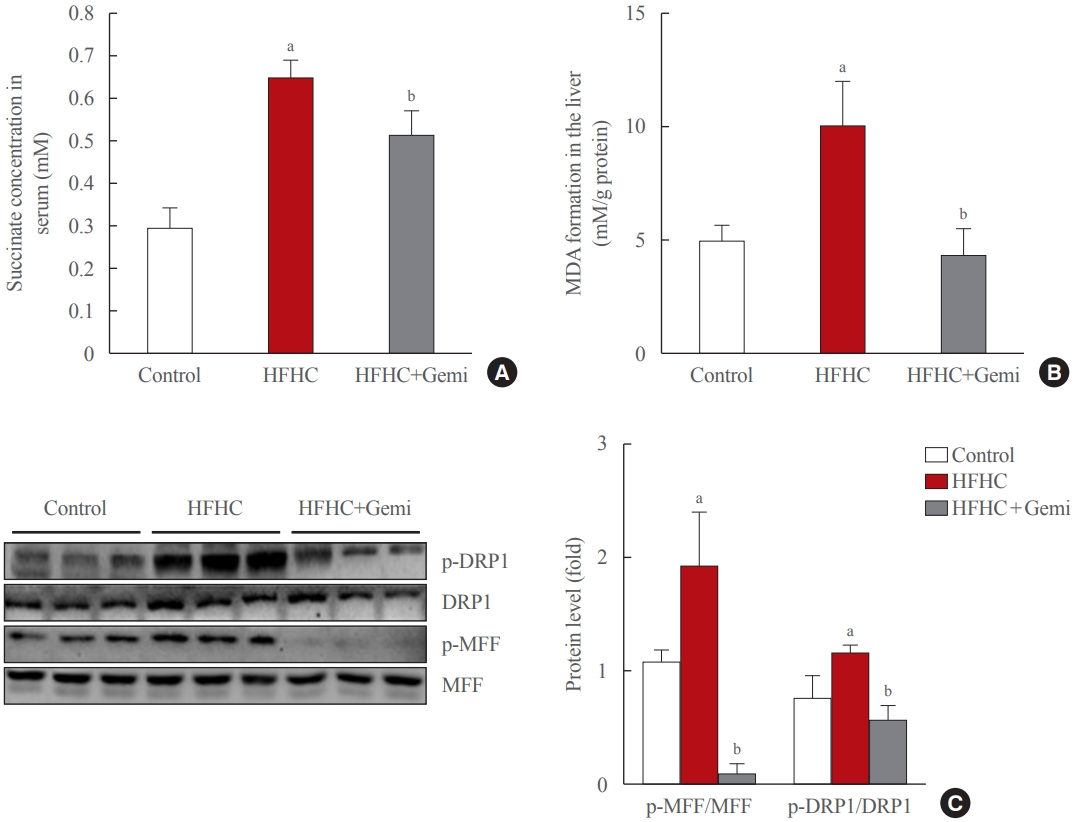
- Gemigliptin protected the liver from oxidative stress and mitochondrial fission
- Succinate concentrations in the blood are clearly elevated in inflammatory-related health conditions, including obesity and type 2 diabetes [29-32]. Therefore, we tested the level of succinate in the blood of 8-week treated mice, and our data showed that gemigliptin administration also markedly reduced succinate levels in the serum of the HFHC diet group, which showed significantly higher succinate levels than the control diet group (Fig. 5A). Next, we measured the formation of MDA, a marker of oxidative stress, in liver tissue of 8-week treated mice. The MDA level was higher in the HFHC group than in the control group, and gemigliptin alleviated MDA levels in the co-treatment group (Fig. 5B).
- Western blot results showed that the HFHC diet enhanced the hepatic expression of mitochondrial fission markers (p-DRP1 and p-MFF) and gemigliptin decreased p-DRP1 and p-MFF proteins in the liver after 8 weeks of feeding (Fig. 5C). Thus, our results suggest that the protective effects of gemigliptin on HFHC diet-induced mouse liver fibrosis may involve altered mitochondrial dysfunction, such as mitochondrial fission.
RESULTS
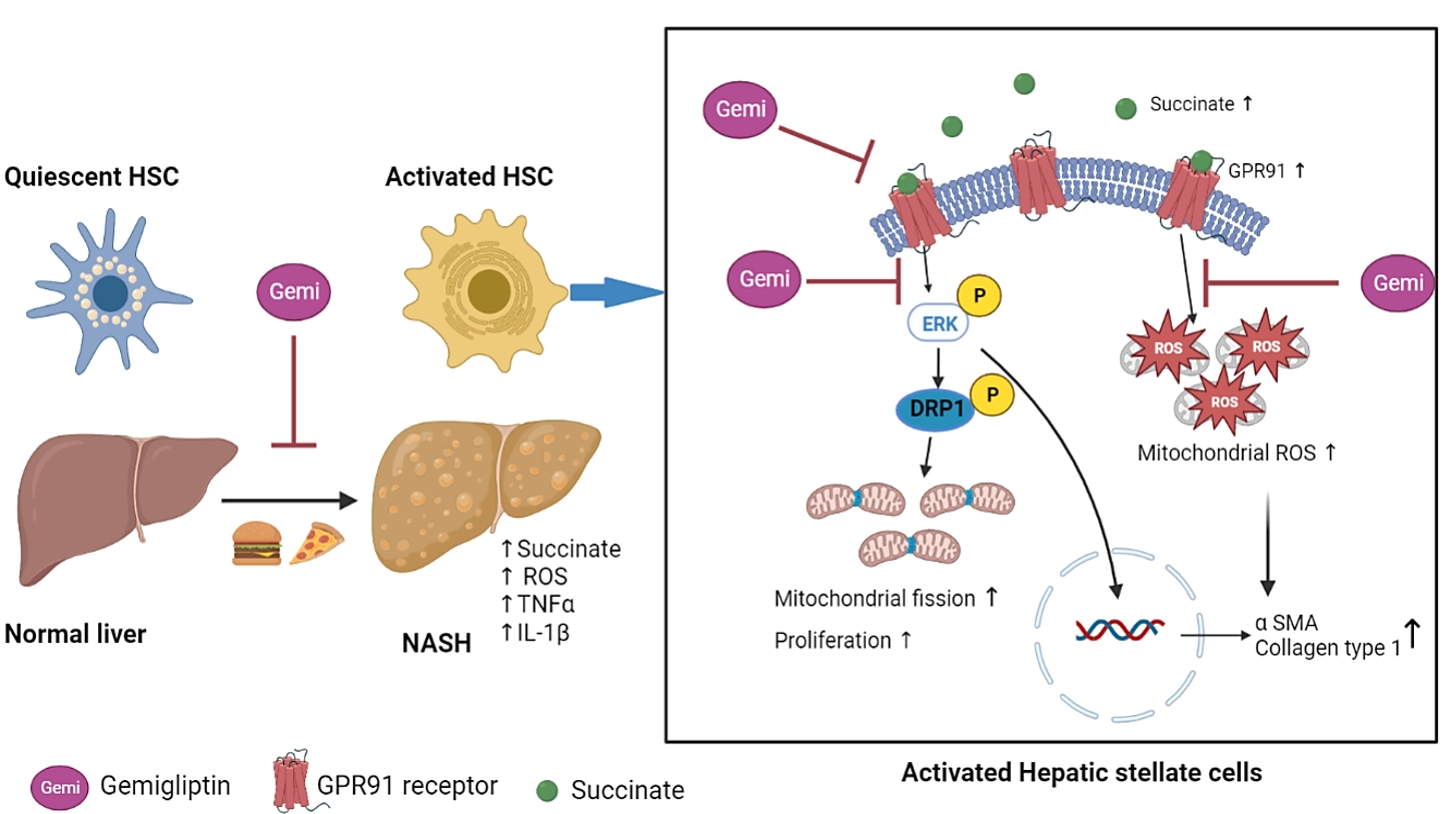
- In the present study, we showed that succinate levels were higher in the serum of HFHC-induced mice and that succinate acted as a signaling molecule to mediate mitochondrial dysfunction and HSC activation via the GPR91 signaling pathway, thus highlighting the pathological impact of succinate in the development and progression of liver fibrosis. Gemigliptin protected the morphological and functional integrity of mitochondria from the succinate insult, thereby attenuating HSC activation in vitro and reducing liver fibrosis in an in vivo HFHC-induced mouse model.
- Although succinate is a citric acid cycle intermediate that is generally synthesized in the mitochondria, it can be released into the extracellular space due to metabolic disturbances and serves as a signaling molecule that regulates cellular responses, including hypoxia, inflammation, oxidative stress, and tumorigenesis [33,34]. High levels of succinate have been detected in the plasma, urine, and cerebral white matter of patients with metabolic diseases [35,36]. In line with these reports, we found elevated succinate levels in the serum of a murine NASH model, suggesting the possibility that released succinate may influence cellular responses in an endocrine manner. Gemigliptin decreased the levels of succinate in circulation in mice treated with both HFHC and gemigliptin. These results suggest that gemigliptin may improve mitochondrial dysfunction by reducing serum succinate levels.
- Mitochondria are important organelles for biosynthetic processes, regulation of stress responses, and cell energy metabolism [37]. Mitochondria act as both a source and a target of intracellular ROS and are involved in the progression of oxidative stress-mediated hepatic fibrosis [38]. In hepatic fibrosis, the activation of HSCs is an important and highly dynamic process involving the synthesis of large amounts of ECM. In vivo, we found that the increase in fibrogenesis markers and inflammatory cytokines may have been correlated with the increase in phosphorylation of DRP1 and MFF, and intracellular ROS in the NASH model. In addition, gemigliptin attenuated liver fibrosis in the treatment group, accompanied by the suppression of mitochondrial fission markers. The succinate insult resulted in increased phosphorylation of DRP-1 (Ser616) and MFF in LX-2 cells, and reduced mitochondrial integrity by inducing mitochondrial fragmentation in HSCs at an early stage. We observed that succinate-induced mitochondrial fission and increased mitochondrial ROS production in LX-2 cells. Interestingly, gemigliptin attenuated mitochondrial fission by inactivating DRP1 and MFF phosphorylation. This effect may contribute to the suppression of mitochondrial ROS production in the HSCs.
- Mitochondrial fission is a highly regulated process that alters the metabolism, proliferation, and apoptosis of cells. MFF has been proposed to act as a DRP1 adaptor to mediate mid-zone fission for mitochondrial biogenesis during cell proliferation [39]. In cardiomyocytes, succinate-mediated GPR91 activation increases ERK1/2 phosphorylation and promotes ERK1/2 translocation to the mitochondria, where it phosphorylates MFF to facilitate DRP1 localization to the mitochondria, thereby contributing to mitochondrial fission [40]. In the liver, HSCs exhibit increased GPR91 expression following succinate stimulation, leading to their activation [8]. GPR91 receptor activation elicits the phosphorylation of downstream signaling molecules, such as ERK, which enhances the expression of nuclear transcription factors and production of α-SMA, thereby stimulating the activation and proliferation of HSCs [8,9]. Taken together, our results show that succinate-induced mitochondrial fission at early time points in HSCs may be associated with the upregulation of GPR91 and increased phosphorylation of the ERK1/2 downstream pathway, which ultimately enhances the proliferation of HSCs. Furthermore, the beneficial effect of gemigliptin on mitochondrial dysfunction also protects against HSC activation by downregulating the succinate-GPR91 signaling pathway.
- Overall, the present study shows that succinate acts as a signaling molecule that mediates mitochondrial dysfunction in parallel with its activation in HSCs. Gemigliptin preserves the morphological integrity of mitochondria and attenuates mitochondrial ROS production by inhibiting DRP1 translocation and MFF phosphorylation. Our work not only provides new insights into the protective effects of gemigliptin against liver fibrosis, but also suggests that suppression of the succinate-GPR91 pathway and modulation of DRP1 phosphorylation represent potential therapeutic strategies for the treatment of liver fibrosis.
DISCUSSION
-
CONFLICTS OF INTEREST
This study was carried out with the support of a research grant from Daewoong Pharmaceuticals, 2017.
-
AUTHOR CONTRIBUTIONS
Conception or design: D.H.C., E.H.C. Acquisition, analysis, or interpretation of data: G.N., S.Y.P., D.V.D. Drafting the work or revising: G.N. Final approval of the manuscript: D.H.C., E.H.C.
Article information
-
Acknowledgements
- This study was carried out with the support of a research grant NRF-2016R1C1B2011968 from the Korean government.



- 1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–18.ArticlePubMedPMC
- 2. Guo J, Friedman SL. Hepatic fibrogenesis. Semin Liver Dis 2007;27:413–26.ArticlePubMed
- 3. Ray I, Mahata SK, De RK. Obesity: an immunometabolic perspective. Front Endocrinol (Lausanne) 2016;7:157.ArticlePubMedPMC
- 4. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest 2013;123:1902–10.ArticlePubMedPMC
- 5. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655–69.ArticlePubMedPMC
- 6. Cho EH. Succinate as a regulator of hepatic stellate cells in liver fibrosis. Front Endocrinol (Lausanne) 2018;9:455.ArticlePubMedPMC
- 7. Li X, Xie L, Qu X, Zhao B, Fu W, Wu B, et al. GPR91, a critical signaling mechanism in modulating pathophysiologic processes in chronic illnesses. FASEB J 2020;34:13091–105.ArticlePubMedPDF
- 8. Li YH, Woo SH, Choi DH, Cho EH. Succinate causes α-SMA production through GPR91 activation in hepatic stellate cells. Biochem Biophys Res Commun 2015;463:853–8.ArticlePubMed
- 9. Park SY, Le CT, Sung KY, Choi DH, Cho EH. Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem Biophys Res Commun 2018;496:673–8.ArticlePubMed
- 10. Cherry C, Thompson B, Saptarshi N, Wu J, Hoh J. 2016: a ‘Mitochondria’ odyssey. Trends Mol Med 2016;22:391–403.ArticlePubMed
- 11. Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol 2014;10:650–8.ArticlePubMedPDF
- 12. Lu B. Mitochondrial dynamics and neurodegeneration; Dordrecht: Springer; 2011. Chapter 2, Relationships between mitochondrial dynamics and bioenergetics. p. 47–68.
- 13. Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to metabolism. Mol Cell 2016;61:683–94.ArticlePubMed
- 14. Archer SL. Mitochondrial dynamics: mitochondrial fission and fusion in human diseases. N Engl J Med 2013;369:2236–51.ArticlePubMed
- 15. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 2010;191:1141–58.ArticlePubMedPMCPDF
- 16. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 2015;57:537–51.ArticlePubMedPMC
- 17. Friedman SL. Liver fibrosis: from bench to bedside. J Hepatol 2003;38 Suppl 1:S38–53.ArticlePubMed
- 18. Fallowfield JA. Therapeutic targets in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 2011;300:G709–15.ArticlePubMed
- 19. Jeong SW. Nonalcoholic fatty liver disease: a drug revolution is coming. Diabetes Metab J 2020;44:640–57.ArticlePubMedPMCPDF
- 20. Balaban YH, Korkusuz P, Simsek H, Gokcan H, Gedikoglu G, Pinar A, et al. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann Hepatol 2007;6:242–50.ArticlePubMed
- 21. Miyazaki M, Kato M, Tanaka K, Tanaka M, Kohjima M, Nakamura K, et al. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol Med Rep 2012;5:729–33.ArticlePubMed
- 22. Thornberry NA, Gallwitz B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract Res Clin Endocrinol Metab 2009;23:479–86.ArticlePubMed
- 23. Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci (Lond) 2005;108:277–92.ArticlePubMedPDF
- 24. Itou M, Kawaguchi T, Taniguchi E, Sata M. Dipeptidyl peptidase-4: a key player in chronic liver disease. World J Gastroenterol 2013;19:2298–306.ArticlePubMedPMC
- 25. Kaji K, Yoshiji H, Ikenaka Y, Noguchi R, Aihara Y, Douhara A, et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J Gastroenterol 2014;49:481–91.ArticlePubMedPDF
- 26. Kawakubo M, Tanaka M, Ochi K, Watanabe A, Saka-Tanaka M, Kanamori Y, et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its antidiabetic effects. Sci Rep 2020;10:983.ArticlePubMedPMCPDF
- 27. Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC. DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J Endocrinol 2013;218:1–11.ArticlePubMed
- 28. Gandhi CR. Oxidative stress and hepatic stellate cells: a paradoxical relationship. Trends Cell Mol Biol 2012;7:1–10.PubMedPMC
- 29. Astiarraga B, Martinez L, Ceperuelo-Mallafre V, Llaurado G, Terron-Puig M, Rodriguez MM, et al. Impaired succinate response to a mixed meal in obesity and type 2 diabetes is normalized after metabolic surgery. Diabetes Care 2020;43:2581–7.ArticlePubMedPMCPDF
- 30. Fernandez-Veledo S, Vendrell J. Gut microbiota-derived succinate: friend or foe in human metabolic diseases? Rev Endocr Metab Disord 2019;20:439–47.ArticlePubMedPMCPDF
- 31. Ceperuelo-Mallafre V, Llaurado G, Keiran N, Benaiges E, Astiarraga B, Martinez L, et al. Preoperative circulating succinate levels as a biomarker for diabetes remission after bariatric surgery. Diabetes Care 2019;42:1956–65.ArticlePubMedPDF
- 32. van Diepen JA, Robben JH, Hooiveld GJ, Carmone C, Alsady M, Boutens L, et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017;60:1304–13.ArticlePubMedPMCPDF
- 33. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013;496:238–42.ArticlePubMedPMCPDF
- 34. Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta 2016;1857:1086–101.ArticlePubMed
- 35. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan Gprotein-coupled receptors. Nature 2004;429:188–93.ArticlePubMedPDF
- 36. Guo Y, Cho SW, Saxena D, Li X. Multifaceted actions of succinate as a signaling transmitter vary with its cellular locations. Endocrinol Metab (Seoul) 2020;35:36–43.ArticlePubMedPMCPDF
- 37. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun 2017;482:426–31.ArticlePubMed
- 38. Ezhilarasan D. Oxidative stress is bane in chronic liver diseases: clinical and experimental perspective. Arab J Gastroenterol 2018;19:56–64.ArticlePubMed
- 39. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021;593:435–9.ArticlePubMedPDF
- 40. Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, et al. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis 2018;9:672.ArticlePubMedPMCPDF
References
Figure & Data
References
Citations
- Improvement effect of gemigliptin on salivary gland dysfunction in exogenous methylglyoxal-injected rats
Woo Kwon Jung, Su-Bin Park, Hwa Young Yu, Junghyun Kim
Heliyon.2024; 10(8): e29362. CrossRef - Gemigliptin, a DPP4 inhibitor, ameliorates nonalcoholic steatohepatitis through AMP-activated protein kinase-independent and ULK1-mediated autophagy
Youngmi Song, Hyekyung Yang, Juhee Kim, Yoonjin Lee, Sung-Ho Kim, In-Gu Do, Cheol-Young Park
Molecular Metabolism.2023; 78: 101806. CrossRef - DPP-4 Inhibitor in Type 2 Diabetes Mellitus Patient with Non-Alcoholic Fatty Liver Disease: Achieving Two Goals at Once?
Ji Cheol Bae
Endocrinology and Metabolism.2022; 37(6): 858. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite