
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 35(3); 2020 > Article
-
Review ArticleWD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System
-
Yeonjoo Kim
 , Soo-Hyun Kim
, Soo-Hyun Kim -
Endocrinology and Metabolism 2020;35(3):494-506.
DOI: https://doi.org/10.3803/EnM.2020.302
Published online: September 8, 2020
Cell Biology Research Centre, Molecular and Clinical Sciences Research Institute, St. George’s, University of London, London, UK
- Corresponding author: Soo-Hyun Kim. Cell Biology Research Centre, Molecular and Clinical Sciences Research Institute, St. George’s, University of London, Cranmer Terrace, London SW17 0RE, UK Tel: +44-208-266-6198, E-mail: skim@sgul.ac.uk
Copyright © 2020 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- WD40-repeat (WDR)-containing proteins constitute an evolutionarily conserved large protein family with a broad range of biological functions. In human proteome, WDR makes up one of the most abundant protein-protein interaction domains. Members of the WDR protein family play important roles in nearly all major cellular signalling pathways. Mutations of WDR proteins have been associated with various human pathologies including neurological disorders, cancer, obesity, ciliopathies and endocrine disorders. This review provides an updated overview of the biological functions of WDR proteins and their mutations found in congenital disorders. We also highlight the significant role of WDR proteins in ciliopathies and endocrine disorders. The new insights may help develop therapeutic approaches targeting WDR motifs.
- WD40-repeat (WDR) refers to a series of loosely conserved structural motifs comprised of approximately 40 amino acids, often terminating in tryptophan (W)-aspartic acid (D). WDR protein family is a large group of proteins commonly possessing the WDR motifs, that are involved in a wide range of important biological processes. Inherited or acquired defects in WDR proteins result in numerous health problems including neurological diseases, ciliopathies, and cancers. In this review, we provide a unique overview and discussion on the molecular mechanisms and functions of WDR proteins, especially focusing on those that have been associated with human congenital disorders and endocrine diseases. Many of the WDR proteins are called different names mainly due to historical reasons. The official gene nomenclature along with full and alternative names of WDR proteins (based on UniProt) discussed in this review are summarised in the Supplemental Table S1. WDR proteins associated with pathological conditions that are not discussed in the main text in detail are summarised in the Supplemental Table S2.
INTRODUCTION
- WDR motif was first described in the β-subunit of a GTP-binding protein transducin complex as a sequence of repeats of 40 to 60 amino acids that begin with glycine and histidine (GH) and end with tryptophan and aspartic acid (WD) dipeptides [1]. WDR is an evolutionarily conserved and highly abundant domain in eukaryotes with nearly 1% of human proteomes consisting of WDR-containing proteins [2]. Most recent protein domain database (SMART, http://smart.embl.de/) predicts that 921 WDR proteins are encoded in humans, 591 in Mus musculus and 212 in Drosophila melanogaster. WDR proteins are rarely present in prokaryotes [2]. Each WDR protein can have 4 to 16 copies of WDRs forming seven or more bladed beta-propeller folds [3], which can provide three structural surfaces (top, side, and bottom region of propeller) available to interact with other binding partners [2]. Based on these structural features, it is suggested that WDR proteins could serve as a scaffold that mediates protein-protein or protein-DNA interaction [4]. Since WDRs do not possess any catalytic activity themselves, functional diversity is likely achieved by coordination of multiple binding partners. Mutations in WDR proteins have been reported in several human diseases. Notably, clinically identified mutations of WDR are mostly found on the surface of the protein, presumably interfering their binding interactions with other proteins [1,2,4].
MOLECULAR STRUCTURE OF WDR PROTEINS
- WDR proteins can be primarily defined by their sequence similarity in the WD40-repeat domain. However, a wide range of sequence variation has been found in WD40-repeats, resulting in variable numbers of beta-propeller structures. Variations outside of the WD-repeat domain can also contribute to the multidomain contexts [5]. In fact, although all WDR proteins are structurally related, their molecular functions can be quite distinct. This functional diversity is usually acquired from the additional domains present in the respective WDR proteins. Currently, more than 360 additional domains are reported in WDR proteins [6]. The most commonly found additional domains are shown to be functionally involved with ubiquitylation (e.g., F-box [7], SOCS-box [8], RING-finger [9]), microtubule dynamics (e.g., CTLH domain [10]), phospholipid-binding (e.g., FYVE domain [11]) and endocytic vesicle coating (e.g., clathrin terminal domain [12]). WDR domains are also identified in essential subunits of multiprotein complexes that participate in various signalling pathways regulating DNA repair [13-15], protein degradation [16,17], cell cycle control [18,19], mRNA translation [20,21], cilia assembly and maintenance [22,23], and hormone biosynthesis [24]. Therefore, it is not surprising that WDR proteins play important roles in fundamental cellular functions, such as signal transduction, gene expression, RNA processing, protein synthesis, homeostasis, proliferation, apoptosis, intracellular vesicle trafficking, and cargo recognition [2,25-29].
BIOLOGICAL FUNCTIONS OF WDR PROTEINS
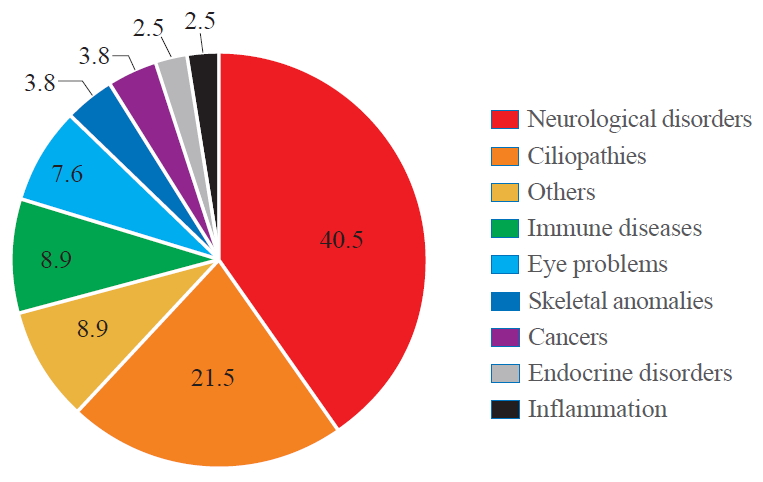
- According to the Online Mendelian Inheritance in Man (OMIM) database (https://omim.org/ ), there is a significant correlation between WDR protein dysfunction and human diseases (Fig. 1). Among 360 WDR genes we assessed, 79 genes were reported to be associated with human pathologies which include neurological disorders (40.5%), ciliopathies (21.5%), immune diseases (8.9%), eye problems (7.6%), skeletal anomalies (3.8%), cancers (3.8%), endocrine disorders (2.5%), inflammation (2.5%), and others including preimplantation embryonic lethality (8.9%).
- Notably, a significant number of WDR proteins have been associated with ciliopathies, a group of genetic disorders resulting from defects in the structure or function of cilia. Cilia are highly conserved microtubule-based hair-like organelles that extend from the plasma membrane of most vertebrate cells. Cilia can broadly be classified into two types, motile and non-motile (primary) cilia that share the principal axoneme structures [30,31]. The axoneme consists of a circular arrangement of nine pairs of microtubules called outer doublets. In addition to the outer doublets, motile cilia contain a pair of microtubules in the centre called inner doublets [32]. This central pair of microtubules is the scaffold of the central pair complex including radial spokes, inner and outer dynein arms and nexin links [33]. The intraflagellar transport (IFT) particles assemble and maintain the cilium by trafficking ciliary proteins within the cilium [22,23]. Two subcomplexes IFT-A and IFT-B, consisting of at least 6 and 13 proteins, respectively, move along the cilium bidirectionally via retrograde (IFT-A) and anterograde (IFT-B) transport. In retrograde transport (from the ciliary tip to the base), IFT-A uses dynein-2 as a motor, whereas IFT-B is powered by kinesin-2 for anterograde movement [34,35]. Many IFT proteins contain protein-protein interaction motifs including WDRs, tetratricopeptide repeats and coiled coils motifs [22], facilitating the interaction and transport of multiple cargos such as tubulin and dynein components [22,36,37]. Primary cilium serves as a regulatory platform and organising centre for many cellular signalling pathways [38] such as Hedgehog [39], receptor tyrosine kinases [40], and G protein-coupled receptors [41], playing critical roles in normal embryonic development and adult homeostasis [42]. Therefore, defects in the formation and function of primary cilia lead to a wide range of health problems [43], including renal dysfunction, retina degeneration, hypogonadism, diabetes, obesity, hearing impairment, craniofacial/skeletal anomalies, cardiovascular defects, and brain malformations [30,44-46], which are collectively termed as ciliopathies.
- WDR proteins associated with neurological disorders
- Mutations of WDR proteins are most frequently associated with neurological disorders (see Table 1 for the full list). PAFAH1B1 (LIS1) is the first WDR protein identified in severe brain malformation called lissencephaly type 1 (also known as classic lissencephaly) characterized by the absence or incomplete development of the cerebral cortex, causing unusually smooth brain surface. Lissencephaly can occur in association with other syndromes such as Miller-Dieker syndrome (MDS) [47,48] or as an isolated lissencephaly sequence (ILS) [49,50]. PAFAH1B1 gene is located in 17p 13.3 which is the most frequently deleted chromosomal region in MDS and ILS patients [51]. So far, it is estimated that 65% of ILS patients have deletions or intragenic mutations of PAFAH1B1 [50]. PAFAH1B1 is a microtubule-associated phosphoprotein and its direct interaction with cytoplasmic dynein heavy change is important for neuronal migration, disruptions of which result in lissencephaly [52,53].
- Mutations in KATNB1 (LIS6) are associated with complex cerebral malformations known as lissencephaly 6 (microcephaly co-existing with lissencephaly) [54,55]. KATNB1 encodes the p80 subunit of katanin, a microtubule-associated ATPase [56,57] which consists of two subunits [56]. While p60 subunit provides the catalytic function, p80 subunit is the regulatory element that targets this protein to centrosomes and maintains the length of microtubules in developing neurons [58,59].
- Mutations in LRRK2 are the most common cause of autosomal dominant Parkinson’s disease [60,61]. To date, more than a hundred mutations of LRRK2 have been identified and six of them are confirmed to be pathogenic [62]. LRRK2 protein binds to the synaptic vesicles and regulates vesicular trafficking by interacting with pre-synaptic proteins such as actin and synapsin [63]. The sequence variation of glycine to arginine at residue 238 (G2385R) which is located between the 5th and 6th WDR domain has been confirmed as a risk factor in the Asian population [64-66]. This mutation modifies LRRK2 protein structure, likely altering its binding affinity to synaptic vesicles and other interactors required for vesicle trafficking [67].
- Cockayne syndrome type A (CSA) is a rare neurodegenerative disorder characterized by complex phenotypes including a growth delay, optic atrophy, deafness, abnormalities in limb and digits, and mental disability [15,68]. According to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ERCC8 ), up to 70 mutations of the ERCC8 gene have been reported in CSA so far. ERCC8 is a subunit of E3 ubiquitin ligase complex [13,14] and interacts with ERCC6 during transcription-coupled nucleotide excision repair [13]. ERCC6, a putative helicase, is recruited by stalled RNA polymerase 2 on the DNA damage site and initiates DNA repair by attracting repair proteins including ERCC8 to the lesion [13,15].
- Triple-A syndrome (AAAS) is a rare autosomal recessive disorder and patients suffer from adrenal insufficiency, achalasia of the oesophagal cardia, alacrimia, and neurological abnormalities affecting the central, peripheral, and autonomic nervous systems [69,70]. Mutations of ALADIN have been found in all AAAS patients, which results in a truncated protein with loss of function [71]. ALADIN protein is normally localised within nuclear pore complexes [18] but mutants of ALADIN are shown to be sequestered in the cytoplasm [72], leading to impaired nuclear transport of proteins that are required to protect the nucleus from oxidative damage [73,74].
- Mutations in WDR4 are reported in patients with microcephaly with severe growth deficiency, seizures, and brain malformations [75,76]. Recent whole-exome sequencing analyses of a family with Galloway-Mowat syndrome (GAMOS) identified a novel mutation of WDR4 [77]. GAMOS present clinically heterogeneous phenotypes which combine renal failure and brain anomalies [78], with additionally associated features including facial dysmorphism, growth retardation and skeletal anomalies [79]. Fibroblast cells derived from patients with GAMOS show defective growth and altered microtubule networks [80]. WDR4 is the human ortholog of yeast Trm82p and forms a complex with N7-methylguanosine tRNA methyltransferase and METTL1, which is essentially required for mRNA translation and stem cell self-renewal and differentiation [20,21]. WDR73 is another WDR protein related to GAMOS [81]. WDR73 is concentrated in the microtubule and interacts with several proteins critical to cell cycle and survival, such as tubulins α/β/γ and Hsp70/ 90 [81].
- WDR proteins and cancer predisposition
- WDR protein PALB2 is a breast and pancreatic cancer susceptibility factor that interacts with BRCA2 and RAD51C [17,37] facilitating their DNA repair function [82]. Cancer-associated PALB2 mutations cause the loss of its binding ability to BRCA2/RAD51C and biallelic mutations of PALB2 are associated with an increased occurrence of childhood cancers [82].
- Another WDR protein FBXW7 is a ubiquitin ligase substrate receptor and the most commonly deregulated ubiquitin/proteasome system (UPS) protein in human cancer [16]. FBXW7 is a tumour suppressor protein that binds to the phosphorylated cyclin E and mediates its degradation by ubiquitination [16,17]. Loss-of-function mutations of FBXW7 result in inappropriate accumulation of cyclin E [17], which is observed in 18% of colorectal cancers, 15% of uterine endometrial carcinoma and 40% of uterine carcinosarcoma [16,17].
- WDR proteins associated with ciliopathies
- To date, mutations in at least 17 different WDR proteins have been identified in ciliopathies (see below and Table 2). Mutations in all components of IFT-A complex—WDR10/IFT122 [83], WDR19/IFT144 [84], WDR35/IFT121 [85], IFT43 [86], IFT140 [87], TTC21B [88]—and a subset of proteins in IFT-B complex—WDR56/IFT80 [89], IFT172 [90], IFT52 [91], IFT81 [92]—have been identified in skeletal ciliopathies. Cranio-ectodermal dysplasia (CED), also known as Sensenbrenner syndrome, is a ciliopathy characterized by craniofacial and skeletal anomalies [93]. So far, four IFT-A proteins are reported to be mutated in CED, namely, WDR10/IFT122, WDR19/IFT144, WDR35/IFT121, and IFT43 [87,94-97]. Mutations in WDR19/IFT144 have also been identified in patients with Jeune syndrome, also known as asphyxiating thoracic dystrophy (ATD), presenting short stature, short digits (brachydactyly), and respiratory distress due to insufficient rib bone growth [84]. WDR35/ IFT121 mutations are found in both Jeune syndrome and Short-Rib-Polydactyly syndrome (SRPS) [98]. Mutations in WDR34 and WDR60 are also associated with ATD and SRPS [98,99]. WDR34 and WDR60 are subunits of the dynein-2 complex, comprising the two intermediate chains of dynein-2 which mediates retrograde ciliary transport via IFT-A [100,101]. In addition to WDR34 and WDR60, disruptions in other dynein-2 subunits are also common causes of ATD and SRPS [89-91]. Therefore, patients with CED, ATD, and SRPS share clinical and genetic features [102] and can also be affected in non-skeletal organs including kidney, eye, liver, and heart [99].
- AHI1 encodes a protein called Jouberin which contains seven WDR domains [103]. Recessive mutations of AHI1 underlie Joubert syndrome (JS) characterized by abnormal development of brain structures, including the cerebellar vermis and the brainstem, which resemble the cross-section of a molar tooth in MRI, thus nicknamed as ‘molar tooth malformation’ [103,104]. JS patients show additional distinctive features including ocular coloboma, polycystic kidney and polydactyly, which are collectively referred to as JS-related disorders (JSRD) [105,106]. AHI1 mutation is also associated with a broad range of neurological disorders including schizophrenia [107] and autism [108]. Recent mouse model studies have revealed that Ahi1 is highly expressed in the postnatal brain and interacts with other proteins crucial for neuronal differentiation [109,110].
- WDR proteins in endocrine disorders
- Table 3 lists the WDR proteins involved in endocrine disorders, many of which are often presented as a part of a ciliopathy. Several signalling receptors important in neuro-endocrine functions are shown to localise to primary cilia [111]. They include kisspeptin receptor (KISS1R) [112], type 1 dopaminergic receptor (D1R), beta-2 adrenergic receptor (B2AR) [113], serotonin receptor 6 (5-HT6) [114], and insulin-like growth factor 1 receptor (IGF1R) [115]. It has been suggested that the spatio-temporal distribution and concentration of these receptors on the ciliary membrane surface may provide an additional level of regulation for the signal capacity and specificity of these receptors [116]. Shortening of cilia length and alteration in ciliation frequency can indicate functional disruption of cilia-dependent receptor signalling and protein trafficking, involved in endocrine functions.
- WDR11 is a scaffolding protein required for normal ciliogenesis. Mutations of WDR11 have been identified in congenital isolated hypogonadotropic hypogonadism (CHH), septo-optic dysplasia (SOD), combined pituitary hormone deficiency (CPHD), and pituitary stalk interruption syndrome [117-120]. CHH is defined by the absent or delayed puberty due to defective gonadotrophin-releasing hormone secretion or action. CHH can present with a normal sense of smell (normosmic CHH) or defective sense of smell (hyposmic/anosmic CHH or Kallmann syndrome) [121,122]. CHH patients often show other associated features such as midline defects (cleft lip or palate), deformity of hands and feet, neurosensory hearing loss, and ocular motor abnormalities [123-125]. Previous studies have suggested that CHH, SOD, and CPHD are genetically overlapping conditions [120]. Clinically identified mutations of WDR11 caused defective cilia formation, and targeted disruption of WDR11 in animal models resulted in dysgenesis of multiple organs affected in CHH and Kallmann syndrome [126]. Hedgehog signalling pathway which depends on the normal function of primary cilia is also shown to be disrupted by the loss of WDR11. Based on these findings, it was suggested that CHH and Kallmann syndrome could be considered as a ciliopathy [126]. The endocrine feature is also common in other ciliopathies such as JS and JSRD. Some JS/JSRD patients show growth hormone or thyroid hormone deficiency [127], CPHD [128], and micropenis [129].
- WDR proteins have been associated with obesity. GNB3 is related to childhood obesity and polymorphism of GNB3 is associated with obesity, hypertension, and diabetes type 2 [130-132]. A genome-wide association study identified WDR11 as a novel genetic locus associated with childhood obesity [133]. Siblings sharing a rare variant of WDR11 gene showed obesity with attention deficit hyperactivity disorder [126]. Obesity is, in fact, one of the main features of Bardet-Biedl syndrome (BBS) [134]. BBS is a ciliopathy with a wide spectrum of clinical features including rod-cone dystrophy, polydactyly, hypogonadism in male and renal abnormalities [135]. Homozygous mutation in WDPCP (also called BBS15) is identified in BBS patients with obesity and male hypogonadism [136]. WDPCP is involved in planar polarity effectors (CPLANE) complex required for recruitment of IFT-A proteins during ciliogenesis [137].
- Mutations of TBL1X have been identified in isolated congenital central hypothyroidism [138]. Congenital hypothyroidism (CH) is a thyroid hormone deficiency at birth caused by the impaired function of the thyroid itself (primary CH) or defective stimulation of thyroid gland by a thyroid-stimulating hormone (central or secondary CH) [139]. Central CH can be categorized into two subtypes—isolated thyroid hormone deficiency and CPHD [139]. Isolated thyroid hormone deficiency accounts for 40% of central CH cases [140] and can be caused by mutations in four genes that regulate the thyroid-stimulating hormone biosynthesis, including β subunit of thyroid-stimulating hormone (TSHβ), receptor for thyrotropin-releasing hormone (TRHR), IGSF1 (the regulator for TRHR expression in the pituitary) and TBL1X (an essential subunit of the thyroid hormone receptor corepressor complex) [24,138].
- Although AAAS was described as a neurological disorder previously, the adrenal glands are one of the primarily affected organs [71,141]. About 85% of AAAS patients show adrenocorticotropin hormone resistant adrenal insufficiency due to impaired glucocorticoid secretion [142] and a subsequence adrenal androgen deficiency is also observed [143]. Homozygous deletion of DMXL2 is identified in patients with the polyendocrinepolyneuropathy syndrome (PEPNS) [144]. PEPNS refers to a combined symptom including CHH with hypothyroidism, hypoglycemia, peripheral polyneuropathy, and mental disability [143]. A recent study has shown that gonad specific DMXL2 deletion causes impaired spermatogenesis in males [145].
WDR PROTEINS IN HUMAN DISEASE
- WDR proteins are widely expressed in human tissues and highly conserved in vertebrates (https://www.proteinatlas.org/search/wdr). Thanks to the recent advancements in genome sequencing analysis, many potentially pathogenic variants of WDR proteins have been identified, which can prove to be powerful tools for assigning new functions to the WDR motifs and associated domains. It is possible that WDR proteins with very similar surfaces have common binding partners or similar functions. Mutations in WDR proteins underlie a broad spectrum of human pathologies including neurological disorders, cancer, ciliopathies, and endocrine disorders. These are complex disorders, thus a clear understanding of the clinical phenotypes and comprehensive diagnosis are often challenging. Molecular mechanisms through which WDR proteins are involved in these diverse conditions remain largely unknown. A better understanding of WDR proteins and their interacting partners may offer some clues. The new insights for WDR-related diseases and their underlying mechanisms as provided in this review may help develop therapeutic approaches targeting the common WDR motifs involved.
CONCLUSIONS
SUPPLEMENTARY MATERIAL
-
Acknowledgements
- This work was supported by Medical Research Council (MRC) project grant MR/L020378/1, Global Educational Trust and St. George’s, University of London.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
WDR, WD40-repeat; MIM, Mendelian Inheritance in Man; AD, autosomal dominant; AR, autosomal recessive; UV, ultraviolet light; XLR, X-linked recessive; ADHD, attention deficit hyperactivity disorder; CATIFA, cleft lip, cataract, tooth abnormality, impaired intellectual development, facial dysmorphism, attention-deficit hyperactivity; XLD, X-linked dominant.
- 1. Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci 2001;58:2085–97.ArticlePubMedPDF
- 2. Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci 2010;35:565–74.ArticlePubMed
- 3. Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell 1995;83:1047–58.ArticlePubMed
- 4. Jain BP, Pandey S. WD40 repeat proteins: signalling scaffold with diverse functions. Protein J 2018;37:391–406.ArticlePubMedPDF
- 5. Yu L, Gaitatzes C, Neer E, Smith TF. Thirty-plus functional families from a single motif. Protein Sci 2000;9:2470–6.ArticlePubMedPMC
- 6. Schapira M, Tyers M, Torrent M, Arrowsmith CH. WD40 repeat domain proteins: a novel target class? Nat Rev Drug Discov 2017;16:773–86.ArticlePubMedPMCPDF
- 7. Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S, Piper RC. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Mol Cell 2010;40:433–43.ArticlePubMedPMC
- 8. Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 2012;64:316–23.ArticlePubMed
- 9. Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell 2003;11:1445–56.ArticlePubMed
- 10. Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, et al. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A 2017;114:E9308–17.ArticlePubMedPMC
- 11. Lystad AH, Simonsen A. Phosphoinositide-binding proteins in autophagy. FEBS Lett 2016;590:2454–68.ArticlePubMed
- 12. von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011;146:471–84.ArticlePubMed
- 13. Wang X, Huang Y, Yan M, Li J, Ding C, Jin H, et al. Molecular spectrum of excision repair cross-complementation group 8 gene defects in Chinese patients with Cockayne syndrome type A. Sci Rep 2017;7:13686.ArticlePubMedPMCPDF
- 14. Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, et al. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev 2006;20:1429–34.ArticlePubMedPMC
- 15. van der Weegen Y, Golan-Berman H, Mevissen TET, Apelt K, Gonzalez-Prieto R, Goedhart J, et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat Commun 2020;11:2104.ArticlePubMedPMC
- 16. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 2014;26:455–64.ArticlePubMedPMC
- 17. Siu KT, Rosner MR, Minella AC. An integrated view of cyclin E function and regulation. Cell Cycle 2012;11:57–64.ArticlePubMedPMC
- 18. Cronshaw JM, Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci U S A 2003;100:5823–7.ArticlePubMedPMC
- 19. Kumar A, Sharma P, Gomar-Alba M, Shcheprova Z, Daulny A, Sanmartin T, et al. Daughter-cell-specific modulation of nuclear pore complexes controls cell cycle entry during asymmetric division. Nat Cell Biol 2018;20:432–42.ArticlePubMedPMCPDF
- 20. Alexandrov A, Martzen MR, Phizicky EM. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 2002;8:1253–66.ArticlePubMedPMC
- 21. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol Cell 2018;71:244–55.ArticlePubMedPMC
- 22. Cole DG. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic 2003;4:435–42.ArticlePubMed
- 23. Hao L, Scholey JM. Intraflagellar transport at a glance. J Cell Sci 2009;122(Pt 7):889–92.ArticlePubMedPMC
- 24. Schoenmakers N, Alatzoglou KS, Chatterjee VK, Dattani MT. Recent advances in central congenital hypothyroidism. J Endocrinol 2015;227:R51–71.ArticlePubMedPMC
- 25. van Nocker S, Ludwig P. The WD-repeat protein superfamily in Arabidopsis: conservation and divergence in structure and function. BMC Genomics 2003;4:50.ArticlePubMedPMC
- 26. Feng Y, Zhang C, Luo Q, Wei X, Jiang B, Zhu H, et al. A novel WD-repeat protein, WDR26, inhibits apoptosis of cardiomyocytes induced by oxidative stress. Free Radic Res 2012;46:777–84.ArticlePubMed
- 27. Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 2004;117:527–39.ArticlePubMed
- 28. Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood 2011;117:4056–64.ArticlePubMedPDF
- 29. Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci 2015;128:207–17.ArticlePubMed
- 30. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 2007;8:880–93.ArticlePubMedPDF
- 31. Malicki JJ, Johnson CA. The cilium: cellular antenna and central processing unit. Trends Cell Biol 2017;27:126–40.ArticlePubMedPMC
- 32. Wemmer KA, Marshall WF. Flagellar motility: all pull together. Curr Biol 2004;14:R992–3.ArticlePubMed
- 33. Smith EF, Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton 2004;57:8–17.ArticlePubMedPMC
- 34. Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat Cell Biol 2013;15:1387–97.ArticlePubMedPMCPDF
- 35. Taschner M, Lorentzen E. The intraflagellar transport machinery. Cold Spring Harb Perspect Biol 2016;8:a028092.ArticlePubMedPMC
- 36. Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol 2002;3:813–25.ArticlePubMedPDF
- 37. Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol 2003;19:423–43.ArticlePubMed
- 38. Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science 2006;313:629–33.ArticlePubMed
- 39. Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 2008;11:277–84.ArticlePubMedPDF
- 40. Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol 2012;226:172–84.ArticlePubMed
- 41. Schou KB, Pedersen LB, Christensen ST. Ins and outs of GPCR signaling in primary cilia. EMBO Rep 2015;16:1099–113.ArticlePubMedPMC
- 42. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010;11:331–44.ArticlePubMedPMCPDF
- 43. Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol 2018;6:8.ArticlePubMedPMC
- 44. Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 2009;151C:281–95.ArticlePubMed
- 45. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 2011;26:1039–56.ArticlePubMedPMCPDF
- 46. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011;364:1533–43.ArticlePubMedPMC
- 47. Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet 1991;48:584–94.PubMedPMC
- 48. Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet 2003;72:918–30.ArticlePubMedPMC
- 49. Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet 1997;6:157–64.ArticlePubMed
- 50. Cardoso C, Leventer RJ, Matsumoto N, Kuc JA, Ramocki MB, Mewborn SK, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet 2000;9:3019–28.ArticlePubMed
- 51. Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 1993;270:2838–42.ArticlePubMed
- 52. Sapir T, Cahana A, Seger R, Nekhai S, Reiner O. LIS1 is a microtubule-associated phosphoprotein. Eur J Biochem 1999;265:181–8.ArticlePubMed
- 53. Jimenez-Mateos EM, Wandosell F, Reiner O, Avila J, Gonzalez-Billault C. Binding of microtubule-associated protein 1B to LIS1 affects the interaction between dynein and LIS1. Biochem J 2005;389(Pt 2):333–41.ArticlePubMedPMCPDF
- 54. Yigit G, Wieczorek D, Bogershausen N, Beleggia F, Moller-Hartmann C, Altmuller J, et al. A syndrome of microcephaly, short stature, polysyndactyly, and dental anomalies caused by a homozygous KATNB1 mutation. Am J Med Genet A 2016;170:728–33.ArticlePubMed
- 55. Mishra-Gorur K, Caglayan AO, Schaffer AE, Chabu C, Henegariu O, Vonhoff F, et al. Mutations in KATNB1 cause complex cerebral malformations by disrupting asymmetrically dividing neural progenitors. Neuron 2014;84:1226–39.ArticlePubMedPMC
- 56. McNally KP, Bazirgan OA, McNally FJ. Two domains of p80 katanin regulate microtubule severing and spindle pole targeting by p60 katanin. J Cell Sci 2000;113(Pt 9):1623–33.ArticlePubMedPDF
- 57. McNally FJ, Okawa K, Iwamatsu A, Vale RD. Katanin, the microtubule-severing ATPase, is concentrated at centrosomes. J Cell Sci 1996;109(Pt 3):561–7.ArticlePubMedPDF
- 58. Hartman JJ, Mahr J, McNally K, Okawa K, Iwamatsu A, Thomas S, et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell 1998;93:277–87.ArticlePubMed
- 59. Ahmad FJ, Yu W, McNally FJ, Baas PW. An essential role for katanin in severing microtubules in the neuron. J Cell Biol 1999;145:305–15.ArticlePubMedPMCPDF
- 60. Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res 2008;86:1711–20.ArticlePubMedPMC
- 61. Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012;491:603–7.ArticlePubMedPMCPDF
- 62. Kluss JH, Mamais A, Cookson MR. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem Soc Trans 2019;47:651–61.ArticlePubMedPMCPDF
- 63. Cirnaru MD, Marte A, Belluzzi E, Russo I, Gabrielli M, Longo F, et al. LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front Mol Neurosci 2014;7:49.ArticlePubMedPMC
- 64. Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord 2007;13:89–92.ArticlePubMed
- 65. Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, et al. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 2007;18:273–5.ArticlePubMed
- 66. Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, et al. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet 2007;120:857–63.ArticlePubMedPDF
- 67. Carrion MDP, Marsicano S, Daniele F, Marte A, Pischedda F, Di Cairano E, et al. The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci Rep 2017;7:5377.ArticlePubMedPMCPDF
- 68. Scheibye-Knudsen M, Tseng A, Borch Jensen M, ScheibyeAlsing K, Fang EF, Iyama T, et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc Natl Acad Sci U S A 2016;113:12502–7.ArticlePubMedPMC
- 69. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet 1978;1:1284–6.ArticlePubMed
- 70. Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, von Petrykowski W, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child 1993;68:779–82.ArticlePubMedPMC
- 71. Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, Huebner A. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet 2001;10:283–90.ArticlePubMed
- 72. Huebner A, Kaindl AM, Knobeloch KP, Petzold H, Mann P, Koehler K. The triple A syndrome is due to mutations in ALADIN, a novel member of the nuclear pore complex. Endocr Res 2004;30:891–9.ArticlePubMed
- 73. Hirano M, Furiya Y, Asai H, Yasui A, Ueno S. ALADINI482S causes selective failure of nuclear protein import and hypersensitivity to oxidative stress in triple A syndrome. Proc Natl Acad Sci U S A 2006;103:2298–303.ArticlePubMedPMC
- 74. Storr HL, Kind B, Parfitt DA, Chapple JP, Lorenz M, Koehler K, et al. Deficiency of ferritin heavy-chain nuclear import in triple a syndrome implies nuclear oxidative damage as the primary disease mechanism. Mol Endocrinol 2009;23:2086–94.ArticlePubMedPMCPDF
- 75. Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, et al. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol 2015;16:210.ArticlePubMedPMC
- 76. Trimouille A, Lasseaux E, Barat P, Deiller C, Drunat S, Rooryck C, et al. Further delineation of the phenotype caused by biallelic variants in the WDR4 gene. Clin Genet 2018;93:374–7.ArticlePubMed
- 77. Braun DA, Shril S, Sinha A, Schneider R, Tan W, Ashraf S, et al. Mutations in WDR4 as a new cause of Galloway-Mowat syndrome. Am J Med Genet A 2018;176:2460–5.ArticlePubMedPMC
- 78. Pezzella M, Yeghiazaryan NS, Veggiotti P, Bettinelli A, Giudizioso G, Zara F, et al. Galloway-Mowat syndrome: an early-onset progressive encephalopathy with intractable epilepsy associated to renal impairment. Two novel cases and review of literature. Seizure 2010;19:132–5.ArticlePubMed
- 79. Krishnamurthy S, Rajesh NG, Ramesh A, Zenker M. Infantile nephrotic syndrome with microcephaly and global developmental delay: the Galloway Mowat Syndrome. Indian J Pediatr 2012;79:1087–90.ArticlePubMedPDF
- 80. Jinks RN, Puffenberger EG, Baple E, Harding B, Crino P, Fogo AB, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain 2015;138(Pt 8):2173–90.ArticlePubMedPMCPDF
- 81. Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet 2014;95:637–48.ArticlePubMedPMC
- 82. Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 2007;39:162–4.ArticlePubMedPDF
- 83. Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, et al. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev Biol 2009;325:225–37.ArticlePubMed
- 84. Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet 2011;89:634–43.ArticlePubMedPMC
- 85. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet 2011;88:508–15.ArticlePubMedPMC
- 86. Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet 2011;48:390–5.ArticlePubMed
- 87. Schmidts M, Frank V, Eisenberger T, Al Turki S, Bizet AA, Antony D, et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum Mutat 2013;34:714–24.ArticlePubMedPMC
- 88. Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet 2011;43:189–96.ArticlePubMedPMCPDF
- 89. Cavalcanti DP, Huber C, Sang KH, Baujat G, Collins F, Delezoide AL, et al. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J Med Genet 2011;48:88–92.ArticlePubMed
- 90. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet 2013;93:915–25.ArticlePubMedPMC
- 91. Zhang W, Taylor SP, Nevarez L, Lachman RS, Nickerson DA, Bamshad M, et al. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet 2016;25:4012–20.ArticlePubMedPMC
- 92. Duran I, Taylor SP, Zhang W, Martin J, Forlenza KN, Spiro RP, et al. Destabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a spectrum of short-rib polydactyly syndrome. Sci Rep 2016;6:34232.ArticlePubMedPMCPDF
- 93. Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, et al. Cranioectodermal dysplasia: a probable ciliopathy. Am J Med Genet A 2009;149A:2206–11.ArticlePubMed
- 94. Levin LS, Perrin JC, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr 1977;90:55–61.ArticlePubMed
- 95. Fry AE, Klingenberg C, Matthes J, Heimdal K, Hennekam RC, Pilz DT. Connective tissue involvement in two patients with features of cranioectodermal dysplasia. Am J Med Genet A 2009;149A:2212–5.ArticlePubMed
- 96. Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 2010;87:418–23.ArticlePubMedPMC
- 97. Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, et al. Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 2010;86:949–56.ArticlePubMedPMC
- 98. Huber C, Wu S, Kim AS, Sigaudy S, Sarukhanov A, Serre V, et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am J Hum Genet 2013;93:926–31.ArticlePubMedPMC
- 99. McInerney-Leo AM, Schmidts M, Cortes CR, Leo PJ, Gener B, Courtney AD, et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet 2013;93:515–23.ArticlePubMedPMC
- 100. Tsurumi Y, Hamada Y, Katoh Y, Nakayama K. Interactions of the dynein-2 intermediate chain WDR34 with the light chains are required for ciliary retrograde protein trafficking. Mol Biol Cell 2019;30:658–70.ArticlePubMedPMC
- 101. Asante D, Stevenson NL, Stephens DJ. Subunit composition of the human cytoplasmic dynein-2 complex. J Cell Sci 2014;127(Pt 21):4774–87.ArticlePubMedPMC
- 102. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat 2018;39:152–66.ArticlePubMed
- 103. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 2004;75:979–87.ArticlePubMedPMC
- 104. Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013;12:894–905.ArticlePubMed
- 105. Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 2009;151C:326–40.ArticlePubMedPMC
- 106. Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A 2004;125A:125–34.ArticlePubMed
- 107. Rivero O, Reif A, Sanjuan J, Molto MD, Kittel-Schneider S, Najera C, et al. Impact of the AHI1 gene on the vulnerability to schizophrenia: a case-control association study. PLoS One 2010;5:e12254.ArticlePubMedPMC
- 108. Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, et al. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet 2008;17:3887–96.ArticlePubMedPMCPDF
- 109. Weng L, Lin YF, Li AL, Wang CE, Yan S, Sun M, et al. Loss of Ahi1 affects early development by impairing BM88/Cend1-mediated neuronal differentiation. J Neurosci 2013;33:8172–84.ArticlePubMedPMC
- 110. Xu X, Yang H, Lin YF, Li X, Cape A, Ressler KJ, et al. Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proc Natl Acad Sci U S A 2010;107:19126–31.ArticlePubMedPMC
- 111. Hilgendorf KI, Johnson CT, Jackson PK. The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol 2016;39:84–92.ArticlePubMedPMC
- 112. Koemeter-Cox AI, Sherwood TW, Green JA, Steiner RA, Berbari NF, Yoder BK, et al. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A 2014;111:10335–40.ArticlePubMedPMC
- 113. Leaf A, Von Zastrow M. Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 2015;4:e06996.ArticlePubMedPMC
- 114. Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res 2000;872:271–5.ArticlePubMed
- 115. Higginbotham H, Guo J, Yokota Y, Umberger NL, Su CY, Li J, et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci 2013;16:1000–7.ArticlePubMedPMCPDF
- 116. Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension. Int J Vasc Med 2011;2011:376281.ArticlePubMedPMCPDF
- 117. McCormack SE, Li D, Kim YJ, Lee JY, Kim SH, Rapaport R, et al. Digenic inheritance of PROKR2 and WDR11 mutations in pituitary stalk interruption syndrome. J Clin Endocrinol Metab 2017;102:2501–7.ArticlePubMedPMCPDF
- 118. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 2010;87:465–79.ArticlePubMedPMC
- 119. Izumi Y, Suzuki E, Kanzaki S, Yatsuga S, Kinjo S, Igarashi M, et al. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril 2014;102:1130–6.ArticlePubMed
- 120. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 2012;97:E694–9.ArticlePubMedPMC
- 121. Kim SH. Congenital hypogonadotropic hypogonadism and Kallmann syndrome: past, present, and future. Endocrinol Metab (Seoul) 2015;30:456–66.ArticlePubMedPMC
- 122. Fraietta R, Zylberstejn DS, Esteves SC. Hypogonadotropic hypogonadism revisited. Clinics (Sao Paulo) 2013;68 Suppl 1(Suppl 1):81–8.ArticlePubMedPDF
- 123. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism. Pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015;11:547–64.ArticlePubMedPDF
- 124. Bonomi M, Libri DV, Guizzardi F, Guarducci E, Maiolo E, Pignatti E, et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012;14:49–56.ArticlePubMed
- 125. Brioude F, Bouligand J, Trabado S, Francou B, Salenave S, Kamenicky P, et al. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotypephenotype relationships. Eur J Endocrinol 2010;162:835–51.ArticlePubMed
- 126. Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, et al. WDR11-mediated Hedgehog signaling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep 2018;19:269–89.ArticlePubMed
- 127. Wolf MT, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, et al. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 2007;72:1520–6.ArticlePubMed
- 128. Akcan N, Bas F, Poyrazoglu S, Bundak R. Joubert syndrome with multiple pituitary hormone deficiency. BMJ Case Rep 2019;12:e229016.ArticlePubMedPMC
- 129. Niceta M, Dentici ML, Ciolfi A, Marini R, Barresi S, Lepri FR, et al. Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: a case report and literature review. BMC Pediatr 2020;20:120.ArticlePubMedPMCPDF
- 130. Li HL, Zhang YJ, Chen XP, Luo JQ, Liu SY, Zhang ZL. Association between GNB3 c.825C > T polymorphism and the risk of overweight and obesity: a meta-analysis. Meta Gene 2016;9:18–25.ArticlePubMedPMC
- 131. Rizvi S, Raza ST, Rahman Q, Mahdi F. Role of GNB3, NET, KCNJ11, TCF7L2 and GRL genes single nucleotide polymorphism in the risk prediction of type 2 diabetes mellitus. 3 Biotech 2016;6:255.ArticlePubMedPMCPDF
- 132. Klenke S, Kussmann M, Siffert W. The GNB3 C825T polymorphism as a pharmacogenetic marker in the treatment of hypertension, obesity, and depression. Pharmacogenet Genomics 2011;21:594–606.ArticlePubMed
- 133. Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One 2012;7:e51954.ArticlePubMedPMC
- 134. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 1989;321:1002–9.ArticlePubMed
- 135. Adam MP, Ardinger HH, Pagon RA, Wallace SE. GeneReviews. Seattle: University of Washington, Seattle; 1993-2020 Chapter, Bardet-Biedl syndrome [cited 2020 Jul 28]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363.
- 136. Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 2010;329:1337–40.ArticlePubMedPMC
- 137. Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet 2016;48:648–56.ArticlePubMedPMCPDF
- 138. Heinen CA, Losekoot M, Sun Y, Watson PJ, Fairall L, Joustra SD, et al. Mutations in TBL1X are associated with central hypothyroidism. J Clin Endocrinol Metab 2016;101:4564–73.ArticlePubMedPMC
- 139. Sugisawa C, Takamizawa T, Abe K, Hasegawa T, Shiga K, Sugawara H, et al. Genetics of congenital isolated TSH deficiency: mutation screening of the known causative genes and a literature review. J Clin Endocrinol Metab 2019;104:6229–37.ArticlePubMedPDF
- 140. Zwaveling-Soonawala N, Naafs JC, Verkerk PH, van Trotsenburg ASP. Mortality in children with early-detected congenital central hypothyroidism. J Clin Endocrinol Metab 2018;103:3078–82.ArticlePubMedPDF
- 141. Brooks BP, Kleta R, Caruso RC, Stuart C, Ludlow J, Stratakis CA. Triple-A syndrome with prominent ophthalmic features and a novel mutation in the AAAS gene: a case report. BMC Ophthalmol 2004;4:7.ArticlePubMedPMC
- 142. Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. Endocr Rev 1998;19:828–43.ArticlePubMedPDF
- 143. Roucher-Boulez F, Brac de la Perriere A, Jacquez A, Chau D, Guignat L, Vial C, et al. Triple-A syndrome: a wide spectrum of adrenal dysfunction. Eur J Endocrinol 2018;178:199–207.ArticlePubMed
- 144. Tata B, Huijbregts L, Jacquier S, Csaba Z, Genin E, Meyer V, et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol 2014;12:e1001952.ArticlePubMedPMC
- 145. Gobe C, Elzaiat M, Meunier N, Andre M, Sellem E, Congar P, et al. Dual role of DMXL2 in olfactory information transmission and the first wave of spermatogenesis. PLoS Genet 2019;15:e1007909.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Exosomes Derived from Bone Marrow Mesenchymal Stem Cells Alleviate Rheumatoid Arthritis Symptoms via Shuttling Proteins
Lijun Wang, Fei Li, Liting Wang, Bingxing Wu, Min Du, Hua Xing, Shifeng Pan
Journal of Proteome Research.2024; 23(4): 1298. CrossRef - Structural screens identify candidate human homologs of insect chemoreceptors and cryptic Drosophila gustatory receptor-like proteins
Richard Benton, Nathaniel J Himmel
eLife.2023;[Epub] CrossRef - Changes in protein phosphorylation by insulin administration in the central nervous system of the gastropod mollusk Lymnaea stagnalis
Junko Nakai, Kengo Namiki, Yuki Totani, Shigeki Yasumasu, Teruki Yoshimura, Takashi Aoki, Etsuro Ito
Biophysics and Physicobiology.2023; 20(4): n/a. CrossRef - Unveiling Distinct Proteomic Signatures in Complicated Crohn’s Disease That Could Predict the Disease Course
Laura A. Lucaciu, Radu Seicean, Alina Uifălean, Maria Iacobescu, Cristina A. Iuga, Andrada Seicean
International Journal of Molecular Sciences.2023; 24(23): 16966. CrossRef - Aromatic patterns: Tryptophan aromaticity as a catalyst for the emergence of life and rise of consciousness
Amal Alachkar
Physics of Life Reviews.2022; 42: 93. CrossRef - EML2-S constitutes a new class of proteins that recognizes and regulates the dynamics of tyrosinated microtubules
Takashi Hotta, Thomas S. McAlear, Yang Yue, Takumi Higaki, Sarah E. Haynes, Alexey I. Nesvizhskii, David Sept, Kristen J. Verhey, Susanne Bechstedt, Ryoma Ohi
Current Biology.2022; 32(18): 3898. CrossRef - Susceptibility of craniofacial ciliopathies to oral cancer-A proposed research
G Arun Kumar
Journal of Dental Health, Oral Disorders & Therapy.2022; 13(2): 41. CrossRef - A WDR47 homolog facilitates ciliogenesis by modulating intraflagellar transport
Chun-Xue Song, Xian-Ting Zeng, Wan-Xin Zeng, Rong Liu, Xia-Jing Tong, Qian Li
Journal of Cell Science.2022;[Epub] CrossRef - Biallelic loss-of-function variants in WDR11 are associated with microcephaly and intellectual disability
Natja Haag, Ene-Choo Tan, Matthias Begemann, Lars Buschmann, Florian Kraft, Petra Holschbach, Angeline H. M. Lai, Maggie Brett, Ganeshwaran H. Mochida, Stephanie DiTroia, Lynn Pais, Jennifer E. Neil, Muna Al-Saffar, Laila Bastaki, Christopher A. Walsh, In
European Journal of Human Genetics.2021; 29(11): 1663. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

