
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 37(5); 2022 > Article
-
Original ArticleMiscellaneous DN200434 Inhibits Vascular Smooth Muscle Cell Proliferation and Prevents Neointima Formation in Mice after Carotid Artery Ligation
 Keypoint
Keypoint
Excessive proliferation and migration of vascular smooth muscle cells (VSMCs) contribute to the development of occlusive vascular diseases. Therefore, therapeutic strategies based on blockade of mitochondrial oxidative phosphorylation are considered promising for the treatment of occlusive vascular diseases. The authors found that DN200434, an orally available estrogen receptor-related gamma inverse agonist, inhibited mitochondrial respiration and mammalian target of rapamycin complex 1 activity and consequently suppressed the proliferation and migration of VSMCs and cell cycle progression. Furthermore, DN200434 reduced carotid artery ligation-induced neointima formation in mice. -
Sudeep Kumar1,2*
 , Jonghwa Jin2*, Hyeon Young Park3*, Mi-Jin Kim2, Jungwook Chin4, Sungwoo Lee4, Jina Kim4, Jung-Guk Kim2, Yeon-Kyung Choi1,5, Keun-Gyu Park1,2,3
, Jonghwa Jin2*, Hyeon Young Park3*, Mi-Jin Kim2, Jungwook Chin4, Sungwoo Lee4, Jina Kim4, Jung-Guk Kim2, Yeon-Kyung Choi1,5, Keun-Gyu Park1,2,3 -
Endocrinology and Metabolism 2022;37(5):800-809.
DOI: https://doi.org/10.3803/EnM.2022.1462
Published online: September 28, 2022
1Research Institute of Aging and Metabolism, Kyungpook National University, Daegu, Korea
2Department of Internal Medicine, Kyungpook National University Hospital, School of Medicine, Kyungpook National University, Daegu, Korea
3Department of Biomedical Science, Graduate School, Kyungpook National University, Daegu, Korea
4New Drug Development Center, Daegu Gyeongbuk Medical Innovation Foundation, Daegu, Korea
5Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Korea
- Corresponding authors: Keun-Gyu Park. Department of Internal Medicine, Kyungpook National University Hospital, School of Medicine, Kyungpook National University, 130 Dongdeok-ro, Jung-gu, Daegu 41944, Korea Tel: +82-53-200-5505, Fax: +82-53-426-2046, E-mail: kpark@knu.ac.kr
- Yeon-Kyung Choi. Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, 807 Hoguk-ro, Buk-gu, Daegu 41404, Korea Tel: +82-53-200-3869, Fax: +82-53-200-3870, E-mail: ykchoi@knu.ac.kr
- *These authors contributed equally to this work.
Copyright © 2022 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Excessive proliferation and migration of vascular smooth muscle cells (VSMCs), which contributes to the development of occlusive vascular diseases, requires elevated mitochondrial oxidative phosphorylation to meet the increased requirements for energy and anabolic precursors. Therefore, therapeutic strategies based on blockade of mitochondrial oxidative phosphorylation are considered promising for treatment of occlusive vascular diseases. Here, we investigated whether DN200434, an orally available estrogen receptor-related gamma inverse agonist, inhibits proliferation and migration of VSMCs and neointima formation by suppressing mitochondrial oxidative phosphorylation.
-
Methods
- VSMCs were isolated from the thoracic aortas of 4-week-old Sprague-Dawley rats. Oxidative phosphorylation and the cell cycle were analyzed in fetal bovine serum (FBS)- or platelet-derived growth factor (PDGF)-stimulated VSMCs using a Seahorse XF-24 analyzer and flow cytometry, respectively. A model of neointimal hyperplasia was generated by ligating the left common carotid artery in male C57BL/6J mice.
-
Results
- DN200434 inhibited mitochondrial respiration and mammalian target of rapamycin complex 1 activity and consequently suppressed FBS- or PDGF-stimulated proliferation and migration of VSMCs and cell cycle progression. Furthermore, DN200434 reduced carotid artery ligation-induced neointima formation in mice.
-
Conclusion
- Our data suggest that DN200434 is a therapeutic option to prevent the progression of atherosclerosis.
- Vascular smooth muscle cells (VSMCs) are abundant in the medial layer of arteries and help to maintain vessel tone, the bloodstream, and blood pressure [1]. Abnormal proliferation and migration of VSMCs play an essential role in occlusive vascular diseases, including atherosclerosis and intimal hyperplasia induced by vascular injury [2]. In response to stimuli that directly cause vascular injuries, such as angioplasty, stent insertion, and coronary artery bypass grafting, intimal hyperplasia in arteries is promoted, and injured arteries are prone to develop vascular restenosis [3,4]. Given that VSMC proliferation and migration have a pivotal role in the pathogenesis of occlusive vascular diseases, accumulating studies have focused on discovering pharmacological molecules that inhibit VSMC proliferation and migration [2,5,6].
- Rapidly proliferating VSMCs require not only increased glycolysis but also enhanced oxidative phosphorylation to meet their increased demands for energy and anabolic precursors [7-9]. Platelet-derived growth factor (PDGF)-stimulated VSMCs exhibit increases in basal respiration, maximal respiratory capacity, and adenosine triphosphate (ATP)-linked respiration in mitochondrial respiration analysis [10-12], whereas proliferation of VSMCs is suppressed when mitochondrial oxidative phosphorylation is inhibited [13-16]. Given that anaplerosis of the citric acid cycle, which is maintained mainly by glutaminolysis, is critical in proliferating cells [17], blockade of glutaminolysis in growth factor-stimulated VSMCs is considered a therapeutic strategy to prevent VSMC proliferation and neointima formation [8,10,18].
- Estrogen-related receptor gamma (ERRγ) is expressed in metabolically active organs such as the heart, kidney, pancreas, and skeletal muscle [19]. It positively regulates mitochondrial bioenergetics, oxidative phosphorylation, and cellular energy homeostasis [20,21]. DN200434, an orally available ERRγ inverse agonist, is a newly developed compound [22]. It effectively enhances radioiodine avidity of anaplastic thyroid carcinoma tumors by upregulating the sodium iodide symporter [22]. However, the effects of DN200434 on proliferation of VSMCs and restenosis remain to be elucidated.
- In this study, we investigated whether DN200434 inhibits proliferation and migration of VSMCs and mitochondrial oxidative phosphorylation, and subsequently suppresses neointima formation in a mouse carotid artery ligation model.
INTRODUCTION
- Cell culture
- Rat aortic VSMCs were prepared as described previously [23]. Briefly, rat aortic smooth muscle cells were isolated from 4-week-old male Sprague-Dawley rats (90 to 100 g) and cultured in plastic culture dishes containing low-glucose Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT, USA) supplemented with 20% fetal bovine serum (FBS, Hyclone) for 2 weeks at 37°C in 5% CO2. The medium was changed daily. Cells at passage 4 to 9 were used in experiments.
- Cell counting
- Primary VSMCs were cultured for 18 hours under serumstarved conditions and then incubated for 24 hours in the presence or absence of 10% FBS or platelet-derived growth factor subunit B (PDGF-BB, 20 ng/mL) with or without 10 μM DN200434 (Daegu Gyeongbuk Medical Innovation Foundation, Daegu, Korea). Hemocytometer-based cell counting was performed using trypan blue solution.
- Western blot analysis
- Western blot analysis was performed as previously described [10]. Protein samples were loaded on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred from gels to polyvinylidene difluoride membranes (Millipore, Danvers, MA, USA). The membranes were washed and blocked with 5% skim milk prepared in Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies against 70 kDa ribosomal protein S6 kinase (p70S6K), phospho-p70S6K (T389), eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), phospho-4E-BP1 (T37/46), proliferating cell nuclear antigen (PCNA), and cyclin D1 (Cell Signaling Technology, Danvers, MA, USA), as well as β-actin (Sigma, St. Louis, MO, USA). The blots were rinsed with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). HRP was detected using enhanced chemiluminescence reagent (BioNote, Hwaseong, Korea).
- Migration assay
- For the wound healing assay, VSMCs (1×105 cells per well) were plated into a 6-well plate and serum-starved for 18 hours. An artificial wound (scratch) was generated using a 200 μL pipette tip. Cells were incubated with or without DN200434 (10 μM) for 24 hours in the presence or absence of 10% FBS or PDGF-BB (20 ng/mL). When the wound had closed, cells were fixed in 4% paraformaldehyde and stained with 0.05% crystal violet (Sigma). For the Transwell migration assay, VSMCs (1×104 cells per well) were seeded onto the microporous membrane (8.0 μm) in the upper chamber of the Transwell (Corning Incorporated, Kennebunk, ME, USA). Cells were serum-starved for 18 hours and then incubated with or without DN200434 for 24 hours in the presence or absence of 20% FBS or PDGF-BB (20 ng/mL). Non-migrated cells in the upper chamber were removed using a cotton swab. Cells that had migrated through the membrane to the lower chamber were fixed with methanol and stained with 0.05% crystal violet. Quantification of transwell migration and wound area were performed using Image J software (National Institutes of Health, Bethesda, MD, USA).
- Immunohistochemistry
- Immunohistochemistry (IHC) was performed on formalinfixed, paraffin-embedded tissue sections, as previously described [10]. The sections were incubated with an anti-PCNA (1:1,500, Cell Signaling Technology) or anti-phospho-4E-BP1 (T37/46) (1:50, Cell Signaling Technology) antibody, and then with a HRP-conjugated secondary antibody, followed by staining with diaminobenzidine (liquid DAB+Substrate Chromogen System, Dako, Carpinteria, CA, USA) and counterstaining with Mayer’s hematoxylin (Merck Inc., Darmstadt, Germany) at room temperature. Images were obtained using a light microscope and the cellSens Entry 1.9 imaging system (Olympus Corporation, Tokyo, Japan). IHC images were quantified using ImageJ software (National Institutes of Health).
- Flow cytometric analysis
- The cell cycle of VSMCs treated with 10% FBS or PDGF-BB (20 ng/mL) with or without 10 μM DN200434 was analyzed by flow cytometry. Cells were serum-starved for 6 hours to synchronize them in G1 phase and then incubated with or without DN200434 for 12 hours in the presence or absence of 10% FBS or PDGF-BB (20 ng/mL). Thereafter, cells were washed with ice-cold phosphate-buffered saline, fixed for 2 hours in cold (–20°C) 70% ethanol, and stained for 30 minutes with propidium iodide (PI)/RNase Staining Buffer (BD Biosciences, San Diego, CA, USA) in the dark. Fluorescence emitted by PI-DNA complexes was measured using an Accuri C6 flow cytometer (BD Biosciences).
- Measurement of the oxygen consumption rate
- The oxygen consumption rate (OCR) of VSMCs was measured using a XF-24 Seahorse Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA). VSMCs were starved for 18 hours and incubated with 10% FBS or PDGF-BB (20 ng/mL) with or without DN200434 (10 μM) for 24 hours. XF Calibrant solution was added to each well of a Seahorse XF-24 plate and incubated at 37°C in a non-CO2 incubator for 1 hour. To investigate changes in cell respiration, 1 μM oligomycin (Sigma), 2 μM carbonyl cyanide 3‑chlorophenylhydrazone (Sigma), and 1 μM rotenone (Sigma) were injected at the indicated time points during the measurement. OCR data were obtained automatically following the manufacturer’s protocol.
- Murine model of carotid artery ligation
- To investigate the effect of DN200434 on neointima formation in vivo, the left common carotid artery of male C57BL/6J mice was ligated as previously described [24]. Briefly, the left common carotid artery proximal to the distal bifurcation was ligated with a 5.0 silk suture, and mice were maintained for 28 days. DN200434 (100 mg/kg/day via oral gavage, 5 days per week for 4 weeks) was orally administered prior to scheduled sacrifice. Arteries proximal to the ligation site were analyzed using hematoxylin and eosin (H&E) and an Elastic van Gieson (EVG) staining kit (Abcam, Cambridge, UK). The cross-sectional intimal and medial areas were measured using ImageJ software (National Institutes of Health). The ratio of the intimal area to the medial area was calculated from the mean of these measurements.
- Ethical statement
- All animal procedures were approved by the Institutional Animal Care and Use Committee of Kyungpook National University (KNU2020-0145).
- Statistical analysis
- All values in graphs represent the mean±standard error of the mean. Statistical analysis to assess differences between groups was performed using a one-way analysis of variance and Dunnett’s post-test (GraphPad Prism 8.0, GraphPad, San Diego, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
METHODS
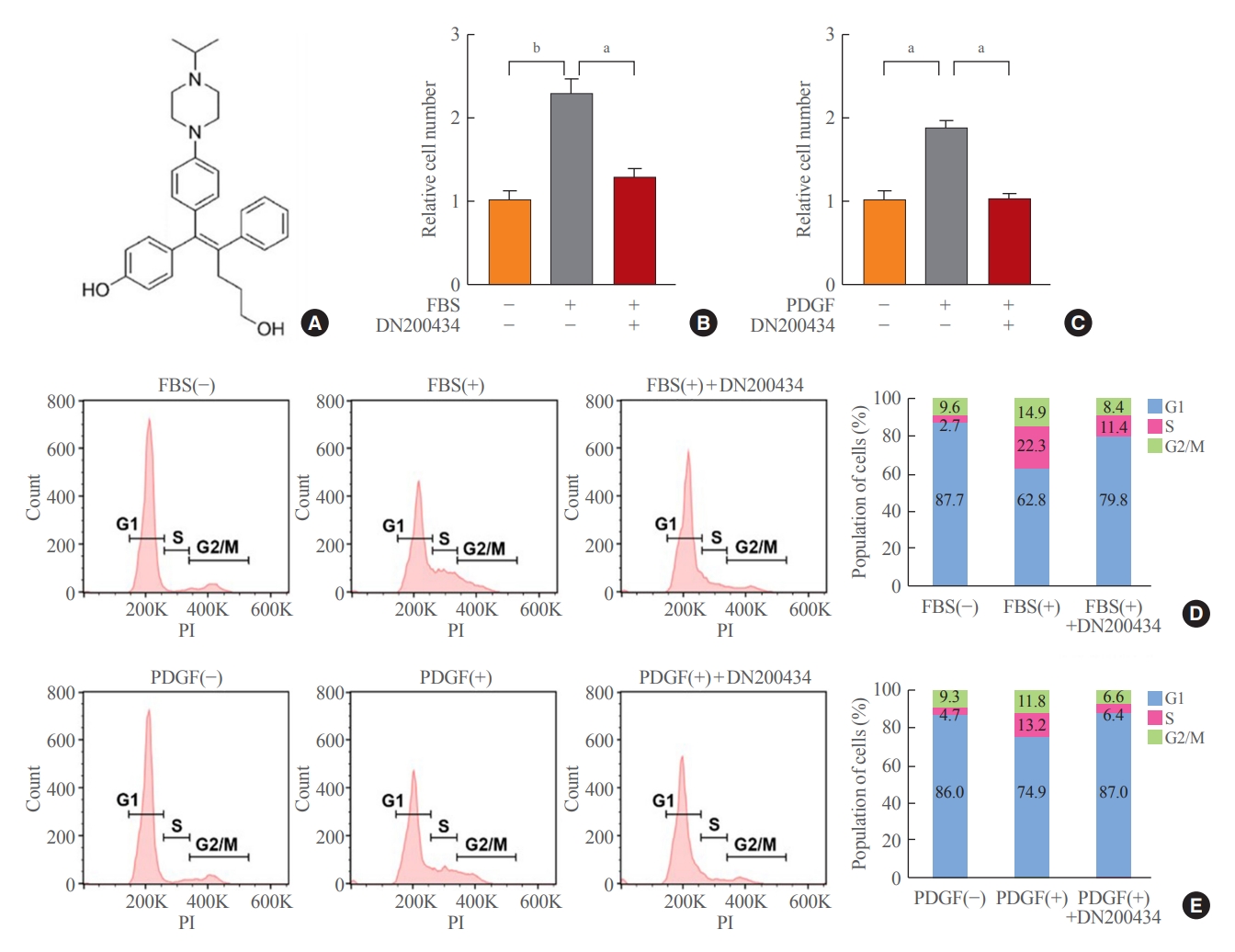
- DN200434 inhibits FBS- or PDGF-stimulated VSMC proliferation and cell cycle progression
- The chemical structure of DN200434 is shown in Fig. 1A. DN20-0434 significantly suppressed FBS- or PDGF-stimulated VSMC proliferation (Fig. 1B, C). In addition, flow cytometric analysis of the cell cycle showed that DN200434 attenuated FBS- or PDGF-stimulated progression from G1 to S phase (Fig. 1D, E).
- DN200434 attenuates mitochondrial respiration in FBSor PDGF-stimulated VSMCs
- Given that oxidative phosphorylation has a role in VSMC proliferation [8,10], we investigated the effects of DN200434 on the OCR of FBS- or PDGF-stimulated VSMCs using an XF analyzer. Consistent with a previous report [8], FBS or PDGF stimulation significantly increased the major parameters of mitochondrial functions, including basal and maximal respiration and ATP-linked respiration, in VSMCs, and these increases were markedly attenuated by DN200434 (Fig. 2).
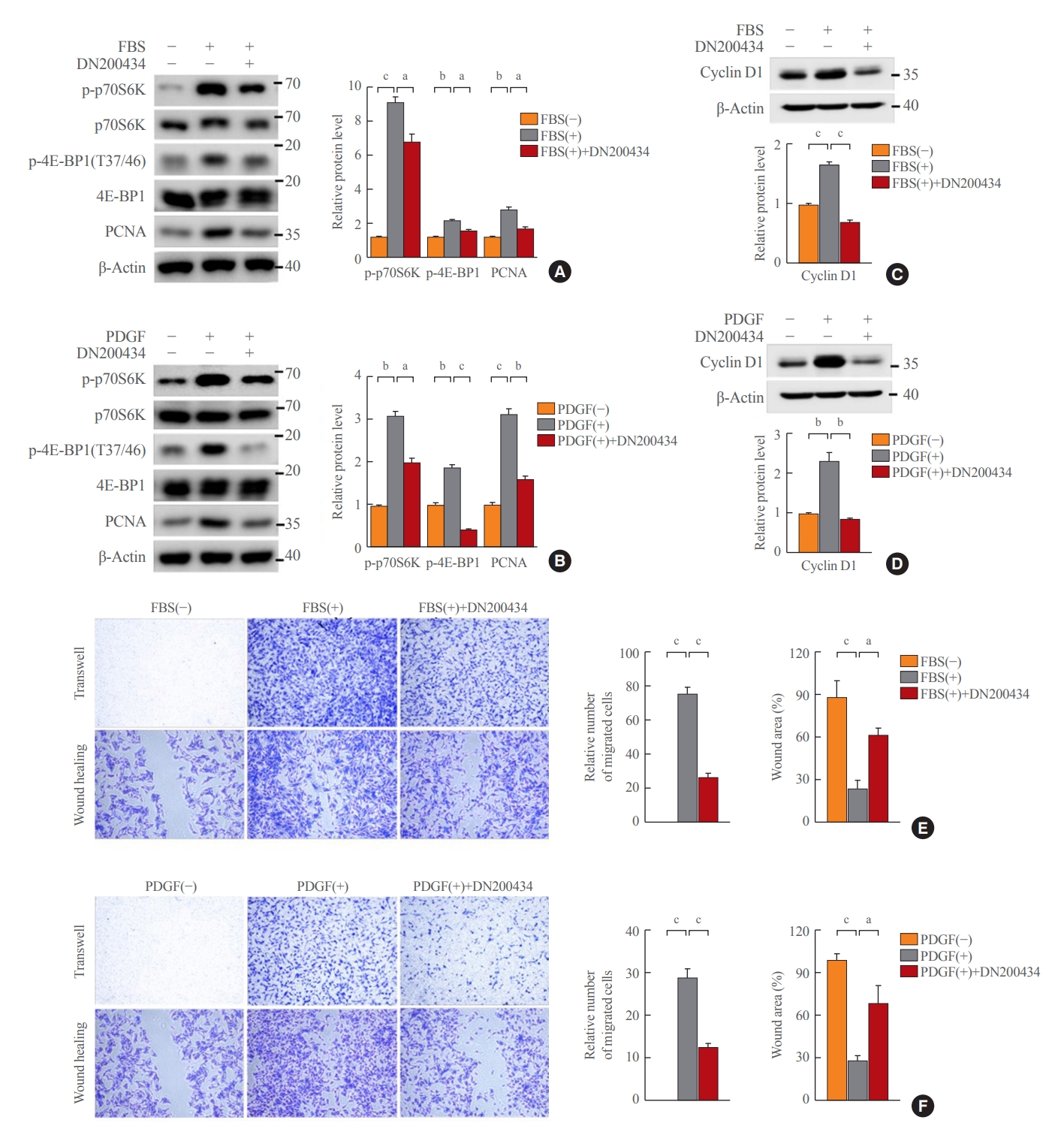
- DN200434 inhibits mTORC1 activity and attenuates cell proliferation and migration in FBS- or PDGF-stimulated VSMCs
- Given that decreased ATP production due to reduced mitochondrial respiration activity inhibits mammalian target of rapamycin complex 1 (mTORC1) activity [25], we investigated the effect of DN200434 on mTORC1 activity in VSMCs. DN200434 inhibited the FBS- or PDGF-induced increases in phosphorylation of p70S6K and 4E-BP1, which are two major downstream targets of mTORC1 (Fig. 3A, B). Furthermore, DN200434 markedly reduced the PCNA level in response to FBS or PDGF stimulation (Fig. 3A, B). Expression of cyclin D1, which controls the transition from G1 to S phase, was increased in FBS- or PDGF-stimulated VSMCs, and these increases were attenuated by DN200434 (Fig. 3C, D). Furthermore, the wound healing and Transwell chamber assays showed that DN200434 attenuated FBS- or PDGF-stimulated migration of VSMCs (Fig. 3E, F).
- DN200434 reduces carotid artery ligation-induced neointimal hyperplasia
- Finally, we investigated the effect of DN200434 on neointima formation using a mouse carotid artery ligation model. H&E and EVG staining of arteries showed severe neointima formation at 4 weeks after carotid artery ligation. Ligation-induced neointimal hyperplasia and the increase in the ratio of intimal area to medial area were significantly attenuated in the DN20-0434-treated group compared with the ligation only group (Fig. 4A, B). Consistent with the effects of DN200434 in vitro, immunohistochemical staining of the carotid artery showed that DN200434 attenuated the increased expression of phospho-4E-BP1 (T37/46) and PCNA in ligation-induced neointimal hyperplasia (Fig. 4C, D).
RESULTS
- In the present study, DN200434 inhibited growth factor-stimulated proliferation, migration, and cell cycle progression of VSMCs by reducing mitochondrial respiration and mTORC1 activity. Moreover, DN200434 significantly attenuated ligationinduced neointimal formation in mice.
- Mitochondrial oxidative phosphorylation is essential in rapidly proliferating VSMCs to meet the increased demands for energy and biomass [26-28]. Our previous study showed that suppression of mitochondrial oxygen consumption via inhibition of glutamine metabolism significantly attenuates VSCM proliferation and neointima formation [8,10]. A growing body of evidence suggests that ERRγ is associated with expression of genes involved in mitochondrial metabolism, including the tricarboxylic acid cycle, fatty acid oxidation, glutamine metabolism, and mitochondrial oxidative respiration [29,30]. A recent study reported that the ERRγ inverse agonist GSK5182 reduces glutamine metabolism and represses glutamine oxidative metabolism in cells exposed to oxidative stress [30]. Consistent with these previous studies, we observed that DN200434 significantly inhibited mitochondrial oxidative phosphorylation in growth factor-stimulated VSMCs.
- Impairment of mitochondrial function negatively regulates mTORC1 activity [8,10]; therefore, reduction of mitochondrial oxidative phosphorylation by DN200434 in VSMCs is critical for suppression of mTORC1 activity. mTORC1 plays a pivotal role in cell growth and proliferation, and activation of mTORC1 is induced by several factors, such as growth factors, ATP, amino acids, and mechanical stimuli [31-36]. mTORC1 activity contributes to aberrant VSMC proliferation and migration, which have a pivotal role in development of atherosclerosis and formation of atherosclerotic plaques; thus, inhibition of mTORC1 is an effective strategy to reduce plaque size and complexity [36,37]. Indeed, mTORC1 activity has been commonly targeted with rapamycin and its analogues to prevent instent restenosis and vascular graft stenosis in clinical practice [38,39]. Furthermore, an mTORC1 inhibitor can induce cell cycle arrest in G1 phase and thus is used in the clinical setting [38,39]. We previously demonstrated the anti-atherosclerotic effects of the glutamine transporter inhibitor V-9302 and the glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) by showing that they decrease mTORC1 activity and mitochondrial oxidative phosphorylation in VSMCs [8,10]. In the present study, we found that DN200434 abrogated growth factor-induced mTORC1 activity and proliferation and migration of VSMCs. Based on our findings that DN200434 decreased the cyclin D1 protein level and induced cell cycle arrest, we conclude that it inhibits proliferation, migration, and cell cycle progression of VSMCs by reducing growth factor-stimulated mTORC1 activity, and consequently has anti-atherosclerotic effects.
- In summary, the results of this study demonstrate that DN200434 suppresses VSMC proliferation and migration by inhibiting mitochondrial oxidative phosphorylation and mTORC1 activity. Given that this newly developed compound can control proliferation of VSMCs, DN200434 is expected to prevent vascular occlusive diseases such as atherosclerosis and restenosis.
DISCUSSION
-
Acknowledgements
- This work was supported by National Research Foundation of Korea (NRF) grants NRF-2017M3A9G7073086, NRF2021R1A2C3005603, NRF-2020R1A5A2017323, and NRF-2022R1A2C1008591, funded by the Ministry of Science and ICT, and by grant HI15C0001 from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTION
Conception or design: S.K., H.Y.P., Y.K.C., K.G.P. Acquisition, analysis, or interpretation of data: S.K., J.J., H.Y.P., M.J.K., J.C., S.L., J.K., J.G.K. Drafting the work or revising: J.J., Y.K.C., K.G.P. Final approval of the manuscript: Y.K.C., K.G.P.
Article information



- 1. Sorokin V, Vickneson K, Kofidis T, Woo CC, Lin XY, Foo R, et al. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front Immunol 2020;11:599415.ArticlePubMedPMC
- 2. Ashino T, Yamamoto M, Numazawa S. Nrf2/Keap1 system regulates vascular smooth muscle cell apoptosis for vascular homeostasis: role in neointimal formation after vascular injury. Sci Rep 2016;6:26291.ArticlePubMedPMCPDF
- 3. Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, et al. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc Sci 2021;2:79–94.ArticlePubMedPMC
- 4. Huang J, Parmacek MS. Modulation of smooth muscle cell phenotype: the other side of the story. Circ Res 2012;111:659–61.ArticlePubMedPMC
- 5. Huang J, Kontos CD. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler Thromb Vasc Biol 2002;22:745–51.ArticlePubMed
- 6. Subbotin VM. Analysis of arterial intimal hyperplasia: review and hypothesis. Theor Biol Med Model 2007;4:41.ArticlePubMedPMCPDF
- 7. Grumbach IM, Nguyen EK. Metabolic stress. Arterioscler Thromb Vasc Biol 2019;39:991–7.ArticlePubMedPMC
- 8. Park HY, Kim MJ, Lee S, Jin J, Lee S, Kim JG, et al. Inhibitory effect of a glutamine antagonist on proliferation and migration of VSMCs via simultaneous attenuation of glycolysis and oxidative phosphorylation. Int J Mol Sci 2021;22:5602.ArticlePubMedPMC
- 9. Perez J, Hill BG, Benavides GA, Dranka BP, Darley-Usmar VM. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem J 2010;428:255–67.ArticlePubMedPMCPDF
- 10. Park HY, Kim MJ, Kim YJ, Lee S, Jin J, Lee S, et al. V-9302 inhibits proliferation and migration of VSMCs, and reduces neointima formation in mice after carotid artery ligation. Biochem Biophys Res Commun 2021;560:45–51.ArticlePubMed
- 11. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol 2013;1:542–51.ArticlePubMedPMC
- 12. Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016;5:e13374.ArticlePubMedPMCPDF
- 13. Lee S, Byun JK, Park M, Kim SW, Lee S, Kim JG, et al. Melatonin inhibits vascular smooth muscle cell proliferation and apoptosis through upregulation of Sestrin2. Exp Ther Med 2020;19:3454–60.ArticlePubMedPMC
- 14. Renner C, Asperger A, Seyffarth A, Meixensberger J, Gebhardt R, Gaunitz F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol Res 2010;32:101–5.Article
- 15. Shen Y, Yang J, Li J, Shi X, Ouyang L, Tian Y, et al. Carnosine inhibits the proliferation of human gastric cancer SGC-7901 cells through both of the mitochondrial respiration and glycolysis pathways. PLoS One 2014;9:e104632.ArticlePubMedPMC
- 16. Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, et al. Mitochondrial respiration: an important therapeutic target in melanoma. PLoS One 2012;7:e40690.ArticlePubMedPMC
- 17. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 2016;126:3313–35.ArticlePubMedPMC
- 18. Osman I, He X, Liu J, Dong K, Wen T, Zhang F, et al. TEAD1 (TEA domain transcription factor 1) promotes smooth muscle cell proliferation through upregulating SLC1A5 (solute carrier family 1 member 5)-mediated glutamine uptake. Circ Res 2019;124:1309–22.ArticlePubMedPMC
- 19. Zhang Z, Teng CT. Interplay between estrogen-related receptor alpha (ERRalpha) and gamma (ERRgamma) on the regulation of ERRalpha gene expression. Mol Cell Endocrinol 2007;264:128–41.PubMed
- 20. Kim JH, Choi YK, Byun JK, Kim MK, Kang YN, Kim SH, et al. Estrogen-related receptor γ is upregulated in liver cancer and its inhibition suppresses liver cancer cell proliferation via induction of p21 and p27. Exp Mol Med 2016;48:e213.ArticlePubMedPMCPDF
- 21. Misra J, Kim DK, Choi HS. ERRγ: a junior orphan with a senior role in metabolism. Trends Endocrinol Metab 2017;28:261–72.ArticlePubMed
- 22. Singh TD, Song J, Kim J, Chin J, Ji HD, Lee JE, et al. A novel orally active inverse agonist of estrogen-related receptor gamma (ERRγ), DN200434, a booster of NIS in anaplastic thyroid cancer. Clin Cancer Res 2019;25:5069–81.ArticlePubMedPDF
- 23. Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev 1979;59:1–61.ArticlePubMed
- 24. Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 1997;17:2238–44.ArticlePubMed
- 25. Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep 2017;21:1331–46.ArticlePubMedPMC
- 26. Alesutan I, Moritz F, Haider T, Shouxuan S, Gollmann-Tepekoylu C, Holfeld J, et al. Impact of β-glycerophosphate on the bioenergetic profile of vascular smooth muscle cells. J Mol Med (Berl) 2020;98:985–97.ArticlePubMedPMCPDF
- 27. Chiong M, Cartes-Saavedra B, Norambuena-Soto I, Mondaca-Ruff D, Morales PE, Garcia-Miguel M, et al. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front Cell Dev Biol 2014;2:72.ArticlePubMedPMC
- 28. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552–63.ArticlePubMedPMC
- 29. Fan W, He N, Lin CS, Wei Z, Hah N, Waizenegger W, et al. ERRγ promotes angiogenesis, mitochondrial biogenesis, and oxidative remodeling in PGC1α/β-deficient muscle. Cell Rep 2018;22:2521–9.ArticlePubMedPMC
- 30. Vernier M, Dufour CR, McGuirk S, Scholtes C, Li X, Bourmeau G, et al. Estrogen-related receptors are targetable ROS sensors. Genes Dev 2020;34:544–59.ArticlePubMedPMC
- 31. Ha JM, Yun SJ, Kim YW, Jin SY, Lee HS, Song SH, et al. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun 2015;464:57–62.ArticlePubMed
- 32. Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol 2017;45:72–82.ArticlePubMedPMC
- 33. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010;328:1172–6.ArticlePubMedPMC
- 34. Cargnello M, Tcherkezian J, Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis 2015;30:169–76.ArticlePubMedPMC
- 35. Bond P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J Int Soc Sports Nutr 2016;13:8.ArticlePubMedPMC
- 36. Zhang S, Lin X, Hou Q, Hu Z, Wang Y, Wang Z. Regulation of mTORC1 by amino acids in mammalian cells: a general picture of recent advances. Anim Nutr 2021;7:1009–23.ArticlePubMedPMC
- 37. Li W, Li Q, Qin L, Ali R, Qyang Y, Tassabehji M, et al. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency. Arterioscler Thromb Vasc Biol 2013;33:1028–35.ArticlePubMed
- 38. Kwon JK, Kim SJ, Kim JH, Lee KM, Chang IH. Dual inhibition by S6K1 and Elf4E is essential for controlling cellular growth and invasion in bladder cancer. Urol Oncol 2014;32:51.Article
- 39. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A 2008;105:17414–9.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Jatrorrhizine inhibits Piezo1 activation and reduces vascular inflammation in endothelial cells
Tianying Hong, Xianmei Pan, Han Xu, Zhijuan Zheng, Lizhen Wen, Jing Li, Mingfeng Xia
Biomedicine & Pharmacotherapy.2023; 163: 114755. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite





