Articles
- Page Path
- HOME > Endocrinol Metab > Volume 36(4); 2021 > Article
-
Review ArticleThyroid The Role of Thyroid Hormone in the Regulation of Cerebellar Development
-
Sumiyasu Ishii*
 , Izuki Amano*, Noriyuki Koibuchi
, Izuki Amano*, Noriyuki Koibuchi -
Endocrinology and Metabolism 2021;36(4):703-716.
DOI: https://doi.org/10.3803/EnM.2021.1150
Published online: August 9, 2021
Department of Integrative Physiology, Gunma University Graduate School of Medicine, Maebashi, Japan
- Corresponding author: Noriyuki Koibuchi. Department of Integrative Physiology, Gunma University Graduate School of Medicine, 3-39-22 Showa-machi, Maebashi 371-8511, Japan, Tel: +81-27-220-7923, Fax: +81-27-220-7926, E-mail: nkoibuch@gunma-u.ac.jp
- * These authors contributed equally to this work.
• Received: June 15, 2021 • Revised: June 30, 2021 • Accepted: July 5, 2021
Copyright © 2021 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- MOLECULAR AND CELLULAR ACTIONS OF TH
- ANIMAL MODEL FOR RESEARCH ON TH ACTION IN THE CEREBELLUM
- DEFECT IN THYROID DYSGENESIS AND THYROID DYSHORMONOGENESIS
- DEFECTS IN TH RECEPTOR ACTION AND FUNCTION
- DEFECTS IN TH TRANSPORT
- DEFECTS IN LOCAL TH METABOLISM
- DEFECTS IN COREGULATORS IN TH ACTION
- CONCLUSIONS
- Article information
- References
ABSTRACT
- The proper organized expression of specific genes in time and space is responsible for the organogenesis of the central nervous system including the cerebellum. The epigenetic regulation of gene expression is tightly regulated by an intrinsic intracellular genetic program, local stimuli such as synaptic inputs and trophic factors, and peripheral stimuli from outside of the brain including hormones. Some hormone receptors are expressed in the cerebellum. Thyroid hormones (THs), among numerous circulating hormones, are well-known major regulators of cerebellar development. In both rodents and human, hypothyroidism during the postnatal developmental period results in abnormal morphogenesis or altered function. THs bind to the thyroid hormone receptors (TRs) in the nuclei and with the help of transcriptional cofactors regulate the transcription of target genes. Gene regulation by TR induces cell proliferation, migration, and differentiation, which are necessary for brain development and plasticity. Thus, the lack of TH action mediators may directly cause aberrant cerebellar development. Various kinds of animal models have been established in a bid to study the mechanism of TH action in the cerebellum. Interestingly, the phenotypes differ greatly depending on the models. Herein we summarize the actions of TH and TR particularly in the developing cerebellum.
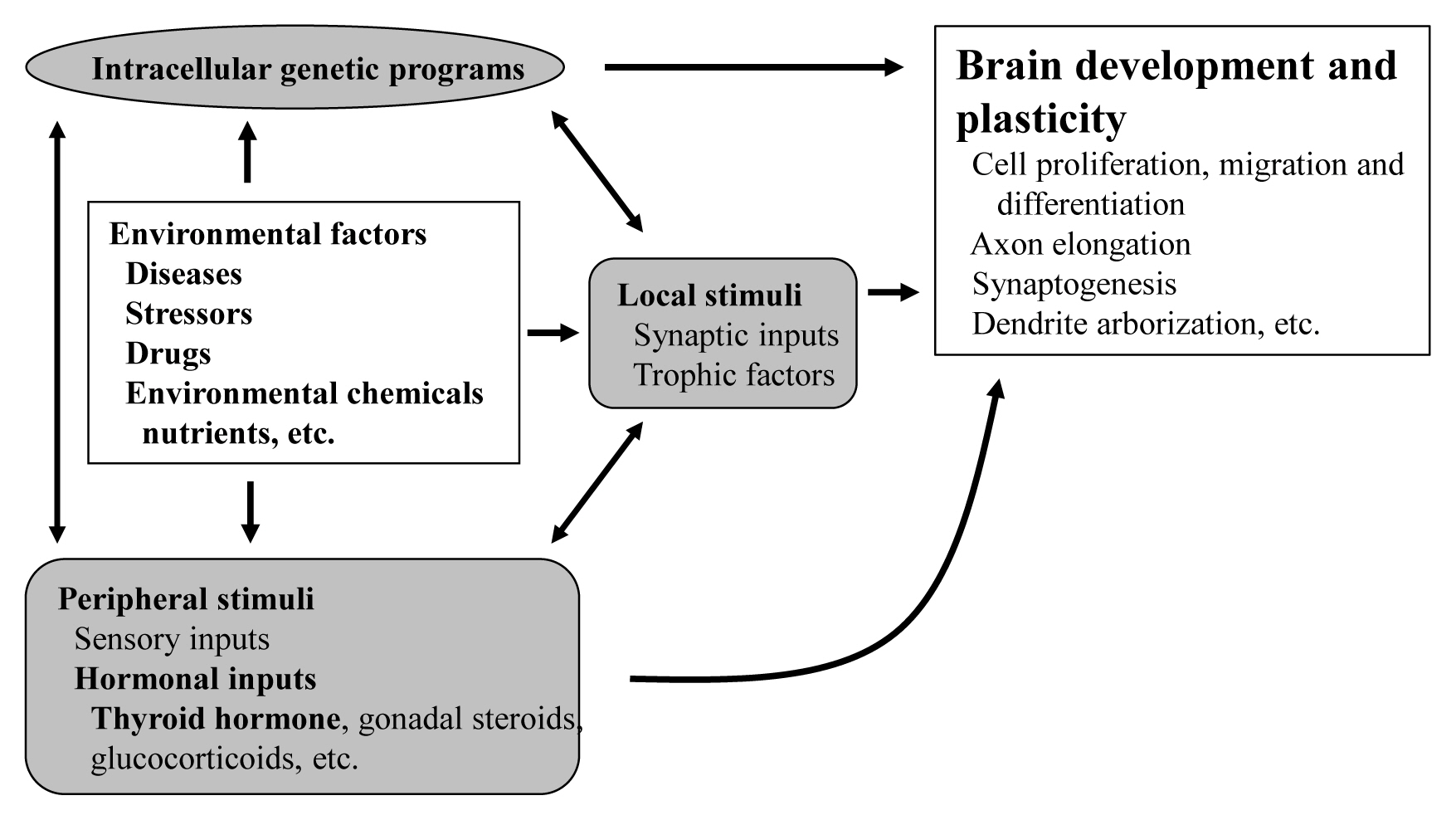
- The proper organized expression of specific genes in time and space is responsible for the organogenesis of central nervous system (CNS) [1]. Developmental defects in the brain are induced by abnormal amount, timing, or area of gene expression. The epigenetic regulation of gene expression is tightly regulated by an intrinsic intracellular genetic program in the neuronal cells themselves, and is also regulated by several stimuli; local and peripheral, from other types of cells (Fig. 1). Local stimuli include synaptic inputs and trophic factors within the brain. Peripheral stimuli from outside of the brain include sensory inputs from the peripheral nervous system and hormonal inputs from endocrine cells. These factors all work together contributing to brain development and plasticity. In addition, environmental factors are important modulators.
- The brain consists of complex interneural networks. The cerebellum is one of few sites in the brain in which the neural network has been extensively studied. The structure of its cortex is well-organized, with a highly specific and uniform arrangement of cells and microcircuitry [2]. Moreover, experimental interventions on the cerebellum are easier in rodents given its postnatal organization. Therefore, the cerebellum is an excellent system for the study of neural development and neural plasticity.
- As peripheral stimuli, hormones produced in the endocrine cells and released into the blood stream significantly influence the development of the brain including the cerebellum. Some hormone receptors are expressed in the cerebellum. However, the role of hormones in the development and plasticity of the cerebellum is not fully understood.
- Among numerous circulating hormones, some small lipophilic hormones including thyroid hormone (TH), gonadal hormones, and glucocorticoids are involved in cerebellar development. These hormones are more capable of crossing the blood-brain barrier (BBB) than peptide hormones because of their chemical properties, although there have been studies incriminating some specific transporters in this process [3]. Receptors for these hormones belong to the nuclear receptor (NR) superfamily and regulate the transcription of the target genes in a ligand-dependent manner [4]. NRs are widely distributed in the CNS with specific expression profiling [5]. Particularly, during cerebellar development, NRs exhibit a temporal and spatial expression pattern [6]. However, the role of NRs during cerebellar development is still under investigation.
- Among these lipophilic hormones, THs are well-known major regulators of cerebellar development. In both rodents [7–9] and humans [10], hypothyroidism during the postnatal developmental period results in abnormal morphogenesis or altered function. This review focuses on the role of TH in cerebellar development. Molecular and cellular actions of TH will be described. Furthermore, cerebellar defects in experimental congenital hypothyroidism (CH) are also summarized.
INTRODUCTION
- THs are synthesized from tyrosine and iodine in the thyroid gland. Thyroid peroxidase and dual oxidase (DUOX) play central roles in the iodine incorporation to tyrosine residues in thyroglobulin [11]. THs include L-triiodothyronine (T3) and L-tetraiodothyronine (thyroxine [T4]). T4 is the main TH produced in the thyroid gland. T3 is produced either directly in the thyroid gland or by deiodination of T4 in the peripheral tissues. Type 2 deiodinase (DIO2) is the dominant enzyme responsible for deiodination in the brain [12]. THs enter the cells largely via membrane transporters such as monocarboxylate transporter 8 (MCT8) [13].
- THs bind to the thyroid hormone receptor (TR) in the nuclei and regulate transcription of the target genes [14]. T3 possesses higher affinity to TR and is regarded as active form. As a member of the NR superfamily, TR contains a DNA-binding domain (DBD) and a ligand-binding domain (LBD). The DBD is highly conserved among multiple NRs, whereas the LBD contains the ligand-dependent activation domain, which is also responsible for dimerization of the NRs. The homology of LBD is relatively high among NRs. The highly variable N-terminal region harbors autonomous activation function.
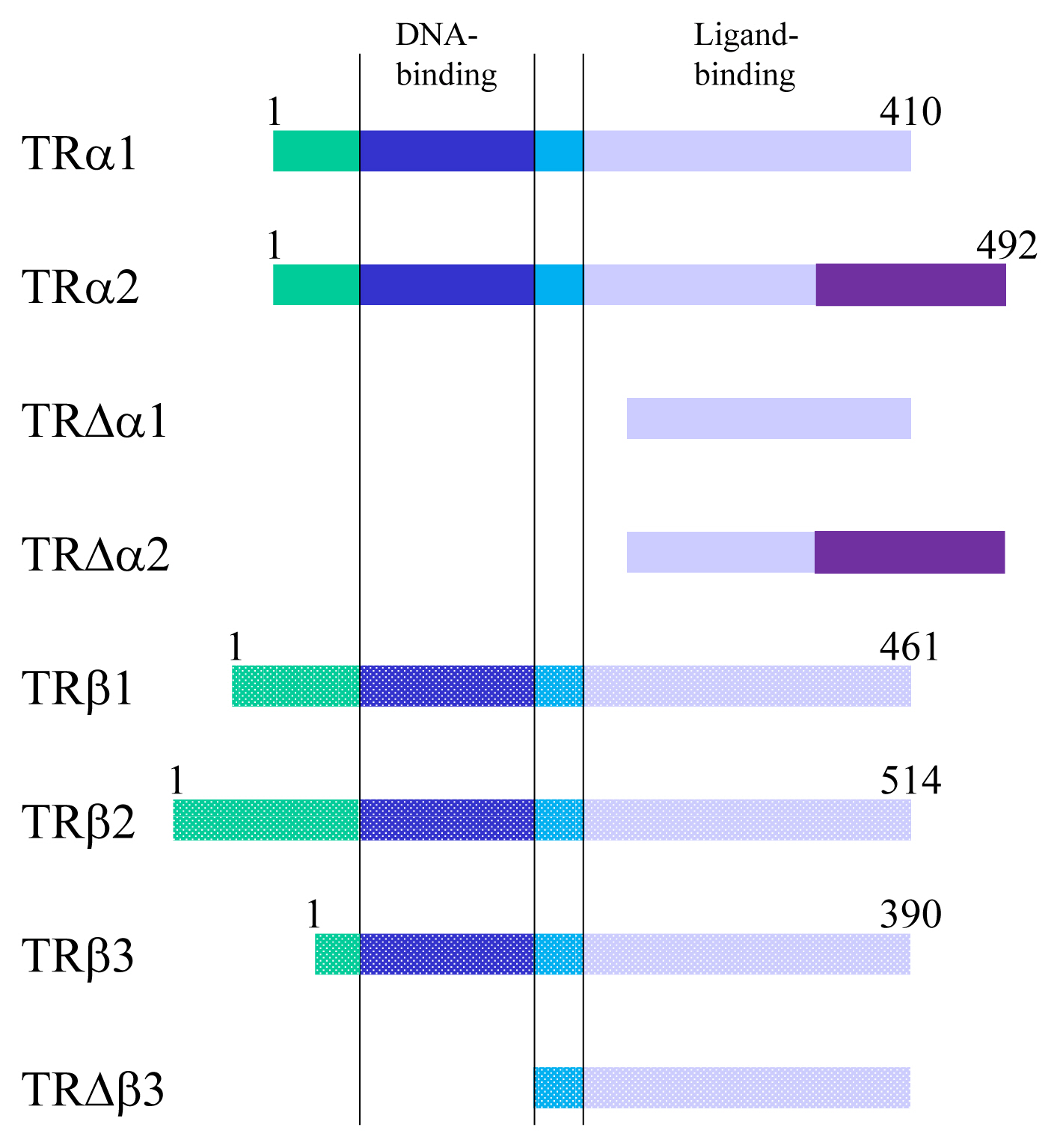
- TRs are encoded by two genes, THRA and THRB, which are located on chromosome 17 and 3 in humans and 11 and 14 in mice [15]. These genes generate several TR isoforms (Fig. 2). Three major isoforms, TRα1, TRβ1, and TRβ2, bind TH and serve as ligand-dependent transcriptional factors. These isoforms are functionally similar; however, their roles are distinguished depending on their distinct expression profiles. TRα2 lacks the capacity to bind TH and works as an endogenous inhibitor of other TRs [16]. In addition, some truncated proteins such as TRΔα1 and TRΔα2 are produced. At least in mice, these two proteins have been shown to be functional transcriptional suppressors in vivo [17]. Furthermore, TRβ3 and its related protein were also reported, although these are regarded as minor isoforms [18].
- TR exerts bi-directional functions similarly to retinoic acid receptor and vitamin D receptor (Fig. 3) [19]. TR binds to specific nucleotide sequences known as TH response element (TRE) on its target genes as a homodimer, or as a heterodimer with retinoid X receptor. Corepressor complexes bind TR and suppress transcription in the absence of a ligand. An addition of TH induces an exchange of cofactors. Corepressor complexes are released from TR and coactivator complexes are recruited, which stimulates transcription. Cofactors including corepressors or coactivators may alter chromatin structure by modulating histone modification or the stability of the basal transcriptional machinery [14,20]. Recently, it was reported that the cofactor exchange does not follow such a canonical “all-or none” switch model in vivo; rather, the expression of TR-target genes is actually regulated by a shift in the relative binding of corepressors and coactivators [21].
- The upregulated target genes are responsible for the cellular effects of THs. These effects include some basic cellular functions such as cell proliferation, migration, and differentiation, which are necessary for brain development and plasticity (Fig. 1). It is suggested that non-genomic action of TH through membranous integrin αvβ3 [22] is responsible for proliferation of cells [23], although TR-target genes responsible for cell proliferation are not yet fully established. TH induces epithelial-mesenchymal transition and stimulates migration by upregulating the zinc finger E-box binding homeobox 1 (ZEB1) gene in squamous cell carcinoma cells [24]. Furthermore, TH plays important roles in the maturation and differentiation of numerous types of cells including neurons and glial cells. Neural differentiation by TH contributes to multiple phenomena such as axon elongation, synaptogenesis, and dendrite arborization. Some examples of TR-target genes responsible for cerebellar development are Purkinje cell protein-2 and myelin basic protein [25]. In addition, the upregulated TR-target genes further induce secondary or tertiary effects. The whole picture of TH-induced cerebellar development remains to be clarified.
MOLECULAR AND CELLULAR ACTIONS OF TH
- TH deficiency during the postnatal period causes CH in humans. Untreated CH causes poor neurodevelopmental outcomes in the children such as mental retardation, cerebellar ataxia, and deafness, together with impaired body growth [26,27]. Thus, the cerebellum has been identified as a TH-sensitive brain region during the developmental age. Various kinds of mice models have been used to assess the effects of TH on the development of the cerebellum (Table 1) [28–52]. These are categorized to drug-induced hypothyroidism, surgical thyroidectomy, or genetic mutation. These models display extensive abnormalities in cerebellar development, resulting in an ataxic phenotype based on morphological or neurophysiological impairment.
ANIMAL MODEL FOR RESEARCH ON TH ACTION IN THE CEREBELLUM
- One of the major causes of primary hypothyroidism is the abnormalities in thyroid gland development (thyroid dysgenesis). The other cause is errors of TH genesis (dyshormonogenesis). The most common CH rodent models are the drug-induced models with anti-thyroid drugs such as propylthiouracil or 2-mercapto-1-methylimidazole (MMI) [53,54]. Nicholson and Altman reported a reduced cerebellar weight; a prolonged cell proliferation in the external granule cell layer (EGL) and retarded EGL disappearance; a retarded cell differentiation in the molecular and internal granule cell layers, and terminal increases in the numbers of granule cells and astrocytes; and a decrease in the numbers of basket cells. They equally reported a myelination delay, and synaptic disconnections among cerebellar neurons and afferent neuronal fibers from other brain regions [55,56]. Legrand [53] and Clos et al. [57] demonstrated a decreased dendrite arborization of Purkinje cells, two-fold-longer primary dendrites, reduction of growth and branching of dendrites, and shorter parallel fibers with fewer synaptic contacts with Purkinje cells. Such impairment of cerebellar Purkinje cell growth was confirmed by ex vivo or in vitro human or rodent models. Moreover, the drug-induced model can enable the study of the effects of mild hypothyroidism on brain development. Mice with mild perinatal hypothyroid had an impaired cerebellar development, and its effect will span across generations.
- In addition to drug-induced hypothyroid animal models, mutations or gene-modifications of several genes that regulate the development of the thyroid gland have been reported as models showing CH. For instance, the paired box 8 (PAX8) gene is required for follicular cells of the thyroid gland for both human and mice [58,59]. Pax8−/− mice showed an organization defect and a reduction of dendrites elaboration that can be rescued by TH treatment [28]. Knockout mice of Duoxa gene (Duoxa−/−), which plays the rate-limiting role of TH synthesis, showed similar morphological changes in the cerebellum at postnatal 2 weeks with severe hypothyroidism. However, cerebellar dysfunction persisted throughout the life although morphological changes had occurred only around the weaning age. Compared to other CH model mice, Duoxa−/− mice may be caused by the combined effect of CH and the functional disruption of the DUOX/DUOXA complex in the brain [29].
DEFECT IN THYROID DYSGENESIS AND THYROID DYSHORMONOGENESIS
- Resistance to thyroid hormone (RTH) is the most common category of impaired sensitivity to TH in humans. RTH is a syndrome characterized by reduced intracellular action of T3 and was first identified as a syndrome of reduced end-organ responsiveness to TH in 1967 [60]. About 85% of families with RTHβ harbor mutations in TRβ [61]. Due to the defects in the feedback regulation of the hypothalamic-pituitary-thyroid axis, patients with RTHβ show high levels of T4 and T3 and normal or high levels of thyroid stimulating hormone (TSH). Patients with RTHβ present with various symptoms. Some patients show hyperthyroidism, while others exhibit hypothyroidism. The diversity of the symptoms can be explained by the various responsiveness to the elevated levels of TH. It was reported that patients RTHβ present a high prevalence of attention deficit disorder and learning disabilities, which are similar to the symptoms of hyperthyroidism [62,63]. However, some patients who harbor mutant TRβ with severe dysfunction have the symptoms of CH. Whereas, RTHα was first discovered in 2012 [64]. Since TRα is not involved in the feedback regulation of the hypothalamic-pituitary-thyroid axis, patients with RTHα tend to have low serum T4, borderline high T3, and lower rT3. Major clinical findings regarding the CNS in patients with RTHα are mental retardation and motor impairment, which are similar to the symptoms of CH [65]. However, much is yet to be known in order to characterize the RTHα mutation in humans.
- RTH model mice, knock-in mice harboring mutant TRs, have been generated. Mice harboring the Δ337T mutant TRβ phenocopy the hypothyroid symptoms of patients with RTHβ carrying the same mutation [30]. In a similar manner to patients with RTHα, mutant TRα knock-in mice also exhibit a hypothyroid phenotype [31]. The neurological phenotypes of the cerebellum of animal models were abnormal cerebellar development, decreased arborization of Purkinje cell dendrites, and impaired locomotor activity. The cerebellar phenotype of RTH animal models is more severe than that of TR knockout animals described below, indicating that the abnormal cerebellar development seen in hypothyroid animals may be induced mainly by unliganded TRs. Furthermore, the effect may not be a result of generalized TH resistance, but may be because of the cerebellar-cell specific action of TH resistance. This hypothesis is supported by studies using animal models expressing dominant-negative TRs in cerebellar cells, showing aberrant cerebellar development [66,67].
- Various TR gene knockout models were also generated; however, these animal models may not always be suitable for studying the action of TH in the cerebellum because of the bi-directional actions of transcriptional regulation of target genes of TR (Fig. 3). Since TR deletion abolishes the repressive action of TR, phenotypes of TR knockout mice are greatly different from those of mice harboring low TH levels. However, TR knockout mice are essential for the study of the role of TR in organ development and function. Another issue that may be considered to generate TR knockout mice is that some introns, such as intron 7 of Thra, have a weak promoter activity. Thus, deleting upstream exons may result in the expression of additional TR-related proteins, which may be limited under normal conditions [68]. As discussed above (Fig. 2), at least three additional TR-related proteins, TRΔα1, TRΔα2, and TRΔβ3, may be generated. Thus, phenotypes of TR knockout mice may result from the combined deletion of a specific TR with overexpression of other TR species. There have also been reports of TRα1-deleted mice showing a limited alterations in behavior and neural circuit [69]. However, except for aberrant maturation of astrocytes, their cerebellar phenotype appeared normal [70]. More strikingly, TRα1 deletion prevented the structural alteration of the cerebellum in hypothyroidism induced by MMI and perchlorate treatment [32]. These results indicate that the abnormal cerebellar phenotype in animals with thyroid dyshormonogenesis may result from the dominant-negative action of unliganded TRα proteins. Interestingly, deleting TRα1 also prevented the structural alteration induced by deleting DIO3 [33]. These results indicate that, liganded TRα1 plays an important role in cerebellar development although the cerebellar phenotype of TRα1 deleted mice is limited. Conversely, TRα2 knockout mice show both hyper- and hypothyroid phenotypes in an organ-specific manner [71]. This may be a result of elevated TRα1 expressions in this mouse. TRα1 expression in the brain is also elevated, but the cerebellar phenotype was unclear. The deletion of both TRα1 and TRα2 also shows only a limited phenotype in the cerebellum. However, besides the cerebellar phenotype, the existence of TRΔα1 and/or TRΔα2 shows altered phenotypes in various organs. When TRα1 and TRα2 are deleted, but expressions of TRΔα1 and TRΔα2 are not inhibited (Thra−/−) [72], their phenotype is more severe than those of mice in which all TRα proteins are deleted (Thra0/0) [17,73]. The decrease in plasma TH levels is greater, and there is a more severe impairment of bone and intestine development. A more limited brain phenotype is observed in TRβ knockout mice, while TRβ1 is widely expressed including in the cerebellum, particularly in Purkinje cells. However, the abnormal brain phenotype seems to be confined to the hypothalamus, and changes in cerebellar phenotype have not been reported [74,75]. Because the function of one receptor cannot be substituted for the other in the case of TRα and β double knockout, their phenotypes are more severe than those of single gene knockout [76,77]. However, there has been no detailed study of the altered brain development in these double knockout mice.
DEFECTS IN TH RECEPTOR ACTION AND FUNCTION
- TH transporters are required to pass the BBB and reach to neurons in the CNS. Two major transmembrane transporters, MCT8 and organic anion transporting polypeptide 1C1 (OATP1C1), are known to transport T3 and T4 specifically. OATP1C1 has a high affinity for T4 and rT3, but a low affinity for T3 [78,79]. MCT8 and OATP1C1 are located in neural cells, the endothelial cells of microvessels, and the choroid plexus [80–82]. In particular, MCT8 is essential for transporting TH through the BBB [34,83]. Solute carrier family 16 member 2 (SLC16A2) mutations, encoding MCT8, cause X-linked Allan-Herndon-Dudley syndrome (AHDS). AHDS is characterized by an altered thyroid function (low T4, high T3, and borderline-increased TSH levels) and TH metabolism and a severely impaired neurodevelopment. Solute carrier organic anion transporter family member 1C1 (SLCO1C1) mutations, encoding OATP1C1, have recently been described to express brain-specific hypothyroidism, severe brain hypometabolism, and juvenile neurodegeneration [84].
- Mice models have been described to assess the function of TH transporters in the CNS. Initially, MCT8 deficient mice were proposed as an AHDS model. Although Mct8−/− mice showed a thyroid profile similar to that of the patients and relatively low levels of T3 in different brain areas, there were no major defects in brain histopathology, nor few behavioral changes [35,36,85]. In particular, Mct8−/− mice revealed no effects on Purkinje cells morphology in the cerebellum [35]. The differences between humans and mice suggest that Mct8−/− mice may have a compensatory mechanism to supply TH to neurons with different factors such as OATP1C1 and DIO2. Interestingly, double knock out of Mct8/Oatp1c1 revealed low uptake of both T3 and T4 into the brain, delayed cerebellar development, and reduced myelination with reduced locomotor activities [37]. Mct8/Dio2 double knockout mice also presented as a model for human MCT8 deficiency, with brain hypothyroidism, alterations in histology, and impaired motor skills [38]. Thus, Mct8 deficiency in mice is compensated by T4 transport through the OATP1C1. This hypothesis is supported by the higher expression of OATP1C1 in rodent BBB compared to that in humans [80,81]. The necessity of MCT8 is also confirmed in the developing chicken cerebellum. The inactivation of MCT8 revealed smaller and less complexed dendritic trees of Purkinje cells, disruption of granule cell precursor proliferation, and post-mitotic granule cell maturation [86]. Recently, the expression pattern of MCT8 appeared to be spatiotemporally changed in the brains of humans and mice [87]. During postnatal development, MCT8 was expressed stably in the endothelial cells of the BBB, choroid plexus epithelial cells, and tanycytes. Conversely, it was robustly detectable in specific brain regions including the cerebellum of young mice and strongly declined with age [87]. These results suggest that spatiotemporal expression patterns of MCT8 contribute to the high TH demands in the developing brain.
DEFECTS IN TH TRANSPORT
- Iodothyronine deiodinase enzymes play important roles for activation and deactivation of TH. In the CNS, DIO2 is predominantly expressed in astrocyte, and converts prohormone T4 to the active hormone T3 [39]. In contrast, DIO3 is mainly expressed in the neuron, and converts T4 to rT3 and T3 to T2 for inactivating THs [88]. It is suggested that the balance between the enzyme activity of DIO2 and DIO3 may determine the local concentration of T3. The balance of DIO2 and DIO3 are known to be controlled dynamically in the rat developing cerebellum [89]. In the second postnatal week, DIO2 activity is upregulated, whereas DIO3 activity is downregulated [90]. These changes correlate with the THs demands, such as myelination and synaptogenesis, in a sensitive period in the mouse cerebellum. Several knockout mice were generated to determine the role of DIO2 and DIO3 in the brain. The Dio2−/− mice was expected to show a similar phenotype to severe global hypothyroid mice. However, neurological functions in Dio2−/− mice were only slightly changed, although the level of T3 was decreased in the cerebellum [40]. Whereas, systemic and astrocyte-specific Dio2−/− mice exhibited increased anxiety and fear memory [39,91]. These reports suggest that compensatory mechanisms protect T3-dependent responses in Dio2−/− mice. This hypothesis was supported by the result of a double knockout of Dio2/Mct8. Mice lacking both Dio2 and Mct8 demonstrated motor skill impairments [38]. In contrast, Dio3−/− mice displayed increased T3-dependent gene expression in the brain suggesting increased of T3 concentration [41,92,93]. Dio3−/− mice exhibited reduced foliation, accelerated disappearance of the EGL, and premature expansion of the molecular layer at juvenile ages [33]. Dio3 knockout mice also exhibited locomotor behavioral abnormalities and impaired ability in descending a vertical pole. Furthermore, double deletion of Dio3 and Tra1 substantially corrected the cerebellar and behavioral phenotypes [33]. Dio3−/− mice also exhibit hyperactivity, decreased anxiety-like behavior, and lack of maternal behavior in female mice [33,94]. These mice models help in understanding that an appropriate balance of DIO2/3 is required in developing brain.
DEFECTS IN LOCAL TH METABOLISM
- The action of TH is mediated by TR and several transcriptional factors. NR coactivators (NCoAs) and corepressors (NCoRs) bind to NRs including TR in a ligand-dependent manner and mediate the transcriptional activities. Nuclear cofactors are known to be expressed in the CNS; however, little is known about their neuronal physiological roles. Recently, reports of genetic variants of nuclear cofactors in CNS-related conditions in humans have begun to emerge [95]. The p160 steroid receptor coactivator (SRC) family comprises three homologous genes: SRC-1 (NCoA1), SRC-2 (NCoA2), and SRC-3 (NCoA3). Heterozygous missense NCoA1 variants found in severely obese individuals impair leptin-mediated proopiomelanocortin (Pomc) reporter activity in humans. Moreover, de novo genetic variants in NCoR1, NCoR2, and histone deacetylase 3 (HDAC3) are found in pediatric patients with intellectual disabilities or autism spectrum disorders (ASD) [42,96–99]. Leptin-induced depolarization of Pomc neurons and Pomc expression were reduced, and food intake and body weight were increased in a knock-in mouse model of a loss of function human variant (SRC-1L1376P) [100]. In the developing rat cerebellum, NCoA1 is predominantly expressed in Purkinje cells and granule cells in the internal granule cell layer. The level of NCoA1 protein in the cerebellum was greatest at P15, when the action of TH may be obvious. The differential expression of SRC-1 may be crucial in mediating the action of TH during cerebellar development [101].
- Mutation each NCoA family showed different phenotypes in mice. Male NCoA1−/− mice had motor learning impairment, while females NCoA2−/− mice had impaired motor coordination. However, NCoA3−/− mice showed no changes on motor skills [43,44]. NCoA2−/− females also showed decreased anxiety, whereas NCoA3−/− female mice had increased anxiety and reduced exploratory activity and impairments in prepulse inhibition [43]. NS-DADm mice, specifically NCoR1/2, abolished their ability to activate HDAC3. These mice showed increased activity, enhanced locomotor coordination, reduced anxiety, impaired social interaction, and impaired spatial learning and recognition memory [42]. Conditional deletion of HDAC3 in mice in generated using nestin-Cre caused abnormal cytoarchitecture of the neocortex and cerebellum, leading to lethality within a day after birth [45]. Another HDAC3 conditional knockout mice using Camk2a-Cre demonstrated a normal organization of cerebellar Purkinje neurons at birth. However, these mice displayed progressive hindlimb paralysis, ataxia, higher numbers of astrocytes, and Purkinje neuron degeneration, which leads to lethality at approximately 6 weeks of age [45,46]. Collectively, both NCoA and NCoR have unique brain regions and gender specific functions.
- Transducin beta-like 1 X-linked (TBL1X) and TBL1X receptor 1 (TBL1XR1), which are the members of HDAC-containing NCoRs complexes, play essential roles in the development of the brain. Targeted sequencing of over 200 genes in more than 10,000 patients with ASD, intellectual disability, seizure, microcephaly, or macrocephaly identified 13 cases carrying disruptive mutations in TBL1XR1 [102]. Moreover, patients with TBL1XR1 mutations display compromised or delayed motor skills [103]. Tbl1xr1−/− mice revealed impaired motor coordination, memory skills, and social activity. Such phenotypes were also described at different levels in patients harboring this mutation [47,103, 104]. TBL1X defects are associated with central hypothyroidism and hearing loss [105,106]. Genome-wide association studies has revealed that TBLX1 is also linked to autism. However, neurodevelopmental defects have not yet been reported in these patients [107]. Further studies are required to identify the role of TBL1XR1 and TBL1X in the developing brain.
- In addition to NcoAs and NcoRs, TR may interact with other NRs that regulate gene expression such as retinoic acid related orphan receptor α (RORα). RORα is strongly expressed in Purkinje cells, and plays a critical role in cerebellar development. Cerebellar phenotype and alteration of neurotrophin expression of natural mutant mouse (staggerer) harboring an RORα mutation is similar to that in the hypothyroid mouse [108], although its thyroid function is normal. This indicates that RORα may be involved in TH-regulated gene expression in the developing cerebellum. In fact, TH regulates RORα expression during the first two postnatal weeks [54,66], indicating that TH may alter gene expression critical for cerebellar development through RORα regulation. Furthermore, RORα augments TR-mediated transcription, whereas staggerer-type mutant RORα does not have such action [108]. Another study showed that RORα may directly interact with TR without binding to TRE. The DBD of RORα may play a role in such an interaction [109]. These results indicate that RORα is required for full TR function in the developing cerebellum. Although there is still little evidence of the mechanism by which transcriptional factors contribute to TH signaling in brain, each transcriptional factor plays an important role in the development of the brain.
DEFECTS IN COREGULATORS IN TH ACTION
- It is established that THs play a pivotal role in cerebellar development. In addition, the molecular mechanisms of the action of TH have been extensively studied. However, the mechanisms through which THs regulate cerebellar development are still under investigation. Further studies including behavioral, neurophysiological, morphological, and molecular aspects are necessary to reveal the whole picture of TH-dependent cerebellar development.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- This study was funded by JSPS KAKENHI Grant No. 21K08569 to Sumiyasu Ishii, Grant-in-Aid for Early-Career Scientists (Grant 19K16486 and 21K15340 to Izuki Amano), and Grant-in-Aid for Scientific Research (B) (Grant 18H03379 to Noriyuki Koibuchi) from the Japan Society for the Promotion of Sciences.
Fig. 2Thyroid hormone receptors and their related proteins generated from THRA or THRB gene. Numerals indicate the number of amino acids. TR, thyroid hormone receptor.

Fig. 3Schematic figure showing the mechanisms of thyroid hormone receptor (TR)-mediated transcription. RXR, retinoid X receptor; TRE, thyroid hormone response element; TF, transcriptional factor.
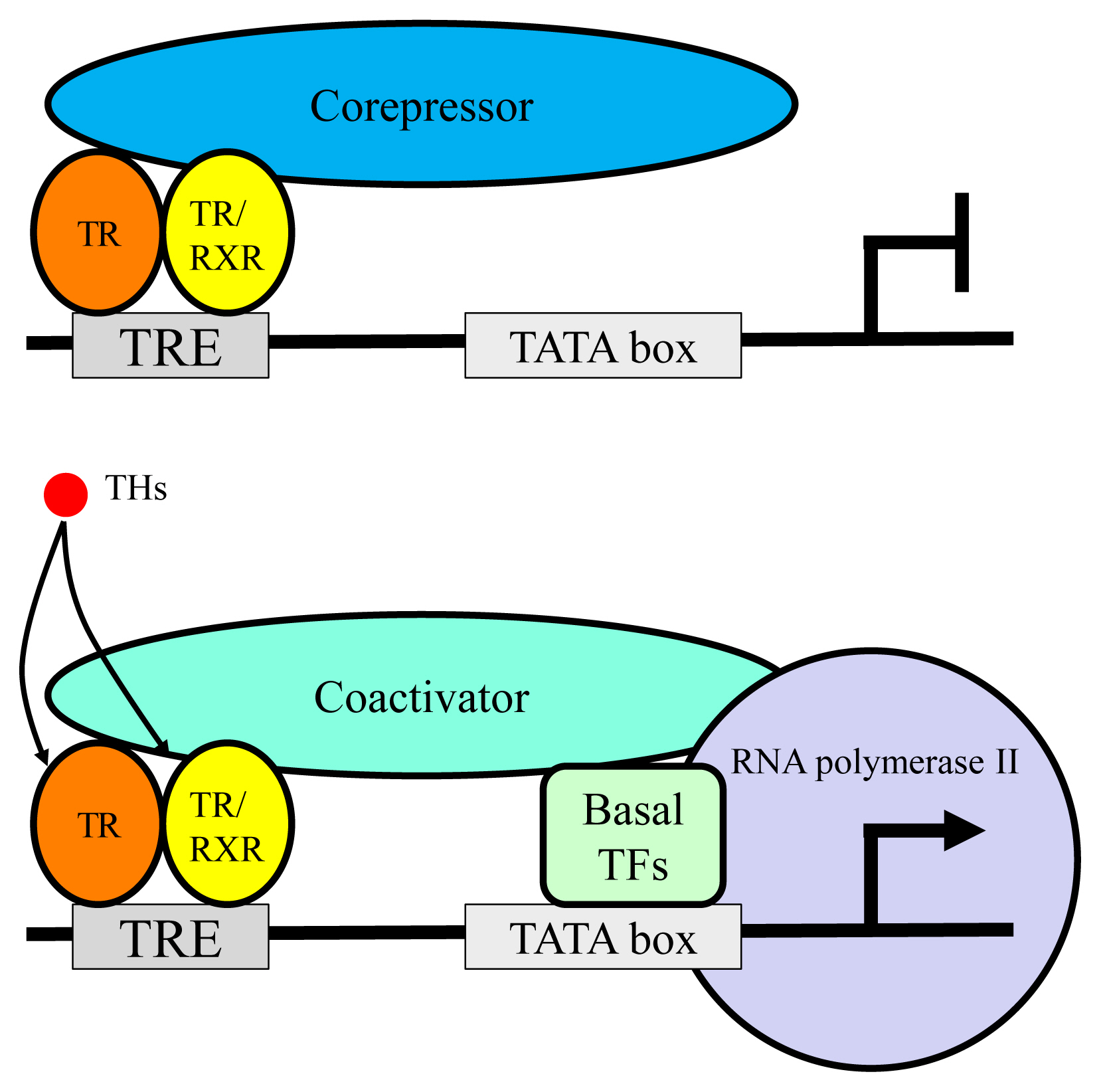
Table 1Representative Phenotype of Animal Models for the Study of the Action of Thyroid Hormone in the Cerebellum
| Species | Category | Gene | Histology | Brain function | Reference | |||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Purkinje cell | Maturation of the external granule cell layer | Locomotor function | Anxiety and sociability | |||||
| Mice | Thyroid gland genesis | Thsr | Abnormal | [48] | ||||
|
|
||||||||
| Mice | Thyroid gland genesis | Pax8 | Abnormal | [28] | ||||
|
|
||||||||
| Mice | TH genesis | Duoxa1/2 | Normal | Abnormal | Abnormal | [29] | ||
|
|
||||||||
| Rat | TH genesis | Tg (missense mutation, known as “rdw rat”) | Abnormal | Abnormal | Abnormal | [49] | ||
|
|
||||||||
| Mice | Receptor | TRα1 | Prevention of hypothyroid phenotype in the cerebellum | [32] | ||||
|
|
||||||||
| Mice | Receptor | TRα1 (mutant knock-in) | Abnormal | Abnormal | Abnormal | Increased anxiety behavior | [31,50,51] | |
|
|
||||||||
| Mice | Receptor | TRβ (mutant knock-in) | Decreased arborization | Abnormal | Abnormal | [30,31] | ||
|
|
||||||||
| Mice | Transporter | Mct8 | Normal | Normal | Normal | Normal | Decreased anxiety related behavior | [34,35,36] |
|
|
||||||||
| Mice | Transporter | Oatp1c1 | Normal | [37] | ||||
|
|
||||||||
| Mice | Transporter | Mct8/Oatp1c1 | Abnormal | [37] | ||||
|
|
||||||||
| Mice | Transporter | Lat2 | Normal | Normal | Normal | Slightly impaired | [52] | |
|
|
||||||||
| Mice | Transporter and deiodinase | Mct8/Dio2 | Abnormal | [38] | ||||
|
|
||||||||
| Mice | Deiodinase | Dio2 | Almost normal | Almost normal | [40] | |||
|
|
||||||||
| Mice | Deiodinase | Dio2 (astrocyte specific) | Anxiety-depression-like behavior | [39] | ||||
|
|
||||||||
| Mice | Deiodinase | Dio3 | Reduced foliation, accelerated disappearance of the external germinal layer, and premature expansion of the molecular layer at juvenile ages | Abnormal | [33] | |||
|
|
||||||||
| Mice | Deiodinase | Dio3 (adult onset) | Increased locomotor activity | [41] | ||||
|
|
||||||||
| Mice | Coactivator | NCoA1 | Delayed maturation | Impaired motor learning in mice | Reduced anxiety | [43,44] | ||
|
|
||||||||
| Mice | Coactivator | NCoA2 | Abnormal in female | Reduced anxiety in female | [43] | |||
|
|
||||||||
| Mice | Coactivator | NCoA3 | Increased anxiety in females | [43] | ||||
|
|
||||||||
| Mice | Corepressor | NCoR1/2 | Increased activity, enhanced locomotor coordination | [42] | ||||
|
|
||||||||
| Mice | Corepressor | HDAC3 (nestin-Cre) | Abnormal cytoarchitecture of the neocortex and cerebellum that leads to lethality within a day after birth | [45] | ||||
|
|
||||||||
| Mice | Corepressor | HDAC3 (CaMKII-Cre) | Purkinje neuron degeneration | Abnormal | [45,46] | |||
|
|
||||||||
| Mice | Corepressor | Tbl1xr1 | Abnormal | [47] | ||||
- 1. Bakken TE, Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, et al. A comprehensive transcriptional map of primate brain development. Nature 2016;535:367–75.ArticlePubMedPMC
- 2. Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, Ding B, et al. Consensus paper: cerebellar development. Cerebellum 2016;15:789–828.ArticlePubMed
- 3. Suzuki T, Abe T. Thyroid hormone transporters in the brain. Cerebellum 2008;7:75–83.ArticlePubMed
- 4. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835–9.ArticlePubMedPMC
- 5. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006;126:789–99.ArticlePubMedPMC
- 6. Qin J, Suh JM, Kim BJ, Yu CT, Tanaka T, Kodama T, et al. The expression pattern of nuclear receptors during cerebellar development. Dev Dyn 2007;236:810–20.ArticlePubMed
- 7. Koibuchi N, Chin WW. Thyroid hormone action and brain development. Trends Endocrinol Metab 2000;11:123–8.ArticlePubMed
- 8. Koibuchi N, Jingu H, Iwasaki T, Chin WW. Current perspectives on the role of thyroid hormone in growth and development of cerebellum. Cerebellum 2003;2:279–89.ArticlePubMed
- 9. Koibuchi N. The role of thyroid hormone on functional organization in the cerebellum. Cerebellum 2013;12:304–6.ArticlePubMed
- 10. Wassner AJ, Brown RS. Hypothyroidism in the newborn period. Curr Opin Endocrinol Diabetes Obes 2013;20:449–54.ArticlePubMedPMC
- 11. Godlewska M, Banga PJ. Thyroid peroxidase as a dual active site enzyme: focus on biosynthesis, hormonogenesis and thyroid disorders of autoimmunity and cancer. Biochimie 2019;160:34–45.ArticlePubMed
- 12. Croteau W, Davey JC, Galton VA, St Germain DL. Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest 1996;98:405–17.ArticlePubMedPMC
- 13. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 2004;74:168–75.ArticlePubMed
- 14. Vella KR, Hollenberg AN. The actions of thyroid hormone signaling in the nucleus. Mol Cell Endocrinol 2017;458:127–35.ArticlePubMedPMC
- 15. Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev 1993;14:184–93.ArticlePubMed
- 16. Koenig RJ, Lazar MA, Hodin RA, Brent GA, Larsen PR, Chin WW, et al. Inhibition of thyroid hormone action by a non-hormone binding c-erbA protein generated by alternative mRNA splicing. Nature 1989;337:659–61.ArticlePubMed
- 17. Gauthier K, Plateroti M, Harvey CB, Williams GR, Weiss RE, Refetoff S, et al. Genetic analysis reveals different functions for the products of the thyroid hormone receptor alpha locus. Mol Cell Biol 2001;21:4748–60.ArticlePubMedPMC
- 18. Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol 2000;20:8329–42.ArticlePubMedPMC
- 19. Yen PM. Classical nuclear hormone receptor activity as a mediator of complex biological responses: a look at health and disease. Best Pract Res Clin Endocrinol Metab 2015;29:517–28.ArticlePubMed
- 20. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 2000;14:121–41.ArticlePubMed
- 21. Shabtai Y, Nagaraj NK, Batmanov K, Cho YW, Guan Y, Jiang C, et al. A coregulator shift, rather than the canonical switch, underlies thyroid hormone action in the liver. Genes Dev 2021;35:367–78.ArticlePubMedPMC
- 22. Davis PJ, Mousa SA, Lin HY. Nongenomic actions of thyroid hormone: the integrin component. Physiol Rev 2021;101:319–52.ArticlePubMed
- 23. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev 2010;31:139–70.ArticlePubMedPMC
- 24. Miro C, Di Cicco E, Ambrosio R, Mancino G, Di Girolamo D, Cicatiello AG, et al. Thyroid hormone induces progression and invasiveness of squamous cell carcinomas by promoting a ZEB-1/E-cadherin switch. Nat Commun 2019;10:5410.ArticlePubMedPMC
- 25. Oppenheimer JH, Schwartz HL. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev 1997;18:462–75.ArticlePubMed
- 26. Hagberg B, Westphal O. Ataxic syndrome in congenital hypothyroidism. Acta Paediatr Scand 1970;59:323–7.ArticlePubMed
- 27. Stenzel D, Huttner WB. Role of maternal thyroid hormones in the developing neocortex and during human evolution. Front Neuroanat 2013;7:19.ArticlePubMedPMC
- 28. Poguet AL, Legrand C, Feng X, Yen PM, Meltzer P, Samarut J, et al. Microarray analysis of knockout mice identifies cyclin D2 as a possible mediator for the action of thyroid hormone during the postnatal development of the cerebellum. Dev Biol 2003;254:188–99.ArticlePubMed
- 29. Amano I, Takatsuru Y, Toya S, Haijima A, Iwasaki T, Grasberger H, et al. Aberrant cerebellar development in mice lacking dual oxidase maturation factors. Thyroid 2016;26:741–52.ArticlePubMedPMC
- 30. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci U S A 2001;98:3998–4003.ArticlePubMedPMC
- 31. Fauquier T, Romero E, Picou F, Chatonnet F, Nguyen XN, Quignodon L, et al. Severe impairment of cerebellum development in mice expressing a dominant-negative mutation inactivating thyroid hormone receptor alpha1 isoform. Dev Biol 2011;356:350–8.ArticlePubMed
- 32. Morte B, Manzano J, Scanlan T, Vennstrom B, Bernal J. Deletion of the thyroid hormone receptor alpha 1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci U S A 2002;99:3985–9.PubMedPMC
- 33. Peeters RP, Hernandez A, Ng L, Ma M, Sharlin DS, Pandey M, et al. Cerebellar abnormalities in mice lacking type 3 deiodinase and partial reversal of phenotype by deletion of thyroid hormone receptor α1. Endocrinology 2013;154:550–61.ArticlePubMed
- 34. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-L-thyronine. Endocrinology 2009;150:2491–6.ArticlePubMedPMC
- 35. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 2007;117:627–35.ArticlePubMedPMC
- 36. Wirth EK, Roth S, Blechschmidt C, Holter SM, Becker L, Racz I, et al. Neuronal 3’,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci 2009;29:9439–49.ArticlePubMedPMC
- 37. Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest 2014;124:1987–99.ArticlePubMedPMC
- 38. Barez-Lopez S, Grijota-Martinez C, Auso E, Fernandez-de Frutos M, Montero-Pedrazuela A, Guadano-Ferraz A. Adult mice lacking Mct8 and Dio2 proteins present alterations in peripheral thyroid hormone levels and severe brain and motor skill impairments. Thyroid 2019;29:1669–82.ArticlePubMed
- 39. Bocco BM, Werneck-de-Castro JP, Oliveira KC, Fernandes GW, Fonseca TL, Nascimento BP, et al. Type 2 deiodinase disruption in astrocytes results in anxiety-depressive-like behavior in male mice. Endocrinology 2016;157:3682–95.ArticlePubMedPMC
- 40. Galton VA, Wood ET, St Germain EA, Withrow CA, Aldrich G, St Germain GM, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology 2007;148:3080–8.ArticlePubMed
- 41. Stohn JP, Martinez ME, St Germain DL, Hernandez A. Adult onset of type 3 deiodinase deficiency in mice alters brain gene expression and increases locomotor activity. Psychoneuroendocrinology 2019;110:104439.ArticlePubMedPMC
- 42. Zhou W, He Y, Rehman AU, Kong Y, Hong S, Ding G, et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus-CA3 projection. Nat Neurosci 2019;22:205–17.ArticlePubMedPMC
- 43. Stashi E, Wang L, Mani SK, York B, O’Malley BW. Research resource: loss of the steroid receptor coactivators confers neurobehavioral consequences. Mol Endocrinol 2013;27:1776–87.ArticlePubMedPMC
- 44. Nishihara E, Yoshida-Komiya H, Chan CS, Liao L, Davis RL, O’Malley BW, et al. SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J Neurosci 2003;23:213–22.ArticlePubMedPMC
- 45. Norwood J, Franklin JM, Sharma D, D’Mello SR. Histone deacetylase 3 is necessary for proper brain development. J Biol Chem 2014;289:34569–82.ArticlePubMedPMC
- 46. Nott A, Cheng J, Gao F, Lin YT, Gjoneska E, Ko T, et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat Neurosci 2016;19:1497–505.ArticlePubMedPMC
- 47. Mastrototaro G, Zaghi M, Massimino L, Moneta M, Mohammadi N, Banfi F, et al. TBL1XR1 ensures balanced neural development through NCOR complex-mediated regulation of the MAPK pathway. Front Cell Dev Biol 2021;9:641410.ArticlePubMedPMC
- 48. Laffan EW, Lisciotto CA, Gapp DA, Weldon DA. Development of rotorod performance in normal and congenitally hypothyroid mutant mice. Behav Neural Biol 1989;52:411–6.ArticlePubMed
- 49. Shimokawa N, Yousefi B, Morioka S, Yamaguchi S, Ohsawa A, Hayashi H, et al. Altered cerebellum development and dopamine distribution in a rat genetic model with congenital hypothyroidism. J Neuroendocrinol 2014;26:164–75.ArticlePubMed
- 50. Venero C, Guadano-Ferraz A, Herrero AI, Nordstrom K, Manzano J, de Escobar GM, et al. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev 2005;19:2152–63.ArticlePubMedPMC
- 51. Wallis K, Sjogren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordstrom K, et al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J Neurosci 2008;28:1904–15.ArticlePubMedPMC
- 52. Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, et al. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J 2011;439:249–55.ArticlePubMed
- 53. Legrand J. Variations, as a function of age, of the response of the cerebellum to the morphogenetic action of the thyroid in rats. Arch Anat Microsc Morphol Exp 1967;56:291–307.PubMed
- 54. Koibuchi N, Yamaoka S, Chin WW. Effect of altered thyroid status on neurotrophin gene expression during postnatal development of the mouse cerebellum. Thyroid 2001;11:205–10.ArticlePubMed
- 55. Balazs R, Kovacs S, Cocks WA, Johnson AL, Eayrs JT. Effect of thyroid hormone on the biochemical maturation of rat brain: postnatal cell formation. Brain Res 1971;25:555–70.ArticlePubMed
- 56. Hajos F, Patel AJ, Balazs R. Effect of thyroid deficiency on the synaptic organization of the rat cerebellar cortex. Brain Res 1973;50:387–401.ArticlePubMed
- 57. Clos J, Crepel F, Legrand C, Legrand J, Rabie A, Vigouroux E. Thyroid physiology during the postnatal period in the rat: a study of the development of thyroid function and of the morphogenetic effects of thyroxine with special reference to cerebellar maturation. Gen Comp Endocrinol 1974;23:178–92.ArticlePubMed
- 58. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet 1998;19:87–90.ArticlePubMed
- 59. Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet 1998;19:83–6.ArticlePubMed
- 60. Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab 1967;27:279–94.PubMed
- 61. Beck-Peccoz P, Mannavola D, Persani L. Syndromes of thyroid hormone resistance. Ann Endocrinol (Paris) 2005;66:264–9.ArticlePubMed
- 62. Hauser P, Zametkin AJ, Martinez P, Vitiello B, Matochik JA, Mixson AJ, et al. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N Engl J Med 1993;328:997–1001.ArticlePubMed
- 63. Brucker-Davis F, Skarulis MC, Grace MB, Benichou J, Hauser P, Wiggs E, et al. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. The National Institutes of Health Prospective Study. Ann Intern Med 1995;123:572–83.ArticlePubMed
- 64. Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med 2012;366:243–9.ArticlePubMed
- 65. Demir K, van Gucht AL, Buyukinan M, Catli G, Ayhan Y, Bas VN, et al. Diverse genotypes and phenotypes of three novel thyroid hormone receptor-α mutations. J Clin Endocrinol Metab 2016;101:2945–54.ArticlePubMed
- 66. Yu L, Iwasaki T, Xu M, Lesmana R, Xiong Y, Shimokawa N, et al. Aberrant cerebellar development of transgenic mice expressing dominant-negative thyroid hormone receptor in cerebellar Purkinje cells. Endocrinology 2015;156:1565–76.ArticlePubMed
- 67. Fauquier T, Chatonnet F, Picou F, Richard S, Fossat N, Aguilera N, et al. Purkinje cells and Bergmann glia are primary targets of the TRα1 thyroid hormone receptor during mouse cerebellum postnatal development. Development 2014;141:166–75.ArticlePubMed
- 68. Chassande O. Do unliganded thyroid hormone receptors have physiological functions? J Mol Endocrinol 2003;31:9–20.ArticlePubMed
- 69. Guadano-Ferraz A, Benavides-Piccione R, Venero C, Lancha C, Vennstrom B, Sandi C, et al. Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol Psychiatry 2003;8:30–8.ArticlePubMedPDF
- 70. Morte B, Manzano J, Scanlan TS, Vennstrom B, Bernal J. Aberrant maturation of astrocytes in thyroid hormone receptor alpha 1 knockout mice reveals an interplay between thyroid hormone receptor isoforms. Endocrinology 2004;145:1386–91.PubMed
- 71. Salto C, Kindblom JM, Johansson C, Wang Z, Gullberg H, Nordstrom K, et al. Ablation of Tralpha2 and a concomitant overexpression of alpha1 yields a mixed hypo- and hyperthyroid phenotype in mice. Mol Endocrinol 2001;15:2115–28.PubMed
- 72. Fraichard A, Chassande O, Plateroti M, Roux JP, Trouillas J, Dehay C, et al. The T3R alpha gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J 1997;16:4412–20.ArticlePubMedPMC
- 73. Macchia PE, Takeuchi Y, Kawai T, Cua K, Gauthier K, Chassande O, et al. Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor alpha. Proc Natl Acad Sci U S A 2001;98:349–54.ArticlePubMed
- 74. Abel ED, Kaulbach HC, Campos-Barros A, Ahima RS, Boers ME, Hashimoto K, et al. Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest 1999;103:271–9.ArticlePubMedPMC
- 75. Sandhofer C, Schwartz HL, Mariash CN, Forrest D, Oppenheimer JH. Beta receptor isoforms are not essential for thyroid hormone-dependent acceleration of PCP-2 and myelin basic protein gene expression in the developing brains of neonatal mice. Mol Cell Endocrinol 1998;137:109–15.ArticlePubMed
- 76. Gothe S, Wang Z, Ng L, Kindblom JM, Barros AC, Ohlsson C, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev 1999;13:1329–41.ArticlePubMedPMC
- 77. Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, et al. Different functions for the thyroid hormone receptors Tralpha and Trbeta in the control of thyroid hormone production and post-natal development. EMBO J 1999;18:623–31.ArticlePubMedPMC
- 78. Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol 2002;16:2283–96.ArticlePubMed
- 79. Tohyama K, Kusuhara H, Sugiyama Y. Involvement of multispecific organic anion transporter, Oatp14 (Slc21a14), in the transport of thyroxine across the blood-brain barrier. Endocrinology 2004;145:4384–91.ArticlePubMed
- 80. Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008;149:6251–61.ArticlePubMed
- 81. Lopez-Espindola D, Garcia-Aldea A, Gomez de la Riva I, Rodriguez-Garcia AM, Salvatore D, Visser TJ, et al. Thyroid hormone availability in the human fetal brain: novel entry pathways and role of radial glia. Brain Struct Funct 2019;224:2103–19.ArticlePubMed
- 82. Schnell C, Shahmoradi A, Wichert SP, Mayerl S, Hagos Y, Heuer H, et al. The multispecific thyroid hormone transporter OATP1C1 mediates cell-specific sulforhodamine 101-labeling of hippocampal astrocytes. Brain Struct Funct 2015;220:193–203.ArticlePubMed
- 83. Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, et al. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell 2017;20:831–43.e5.ArticlePubMedPMC
- 84. Stromme P, Groeneweg S, Lima de Souza EC, Zevenbergen C, Torgersbraten A, Holmgren A, et al. Mutated thyroid hormone transporter OATP1C1 associates with severe brain hypometabolism and juvenile neurodegeneration. Thyroid 2018;28:1406–15.ArticlePubMed
- 85. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 2006;147:4036–43.ArticlePubMed
- 86. Delbaere J, Vancamp P, Van Herck SL, Bourgeois NM, Green MJ, Wingate RJ, et al. MCT8 deficiency in Purkinje cells disrupts embryonic chicken cerebellar development. J Endocrinol 2017;232:259–72.ArticlePubMed
- 87. Wilpert NM, Krueger M, Opitz R, Sebinger D, Paisdzior S, Mages B, et al. Spatiotemporal changes of cerebral monocarboxylate transporter 8 expression. Thyroid 2020;30:1366–83.ArticlePubMed
- 88. Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology 1999;140:784–90.ArticlePubMed
- 89. Kaplan MM, Yaskoski KA. Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J Clin Invest 1981;67:1208–14.ArticlePubMedPMC
- 90. Bates JM, St Germain DL, Galton VA. Expression profiles of the three iodothyronine deiodinases, D1, D2, and D3, in the developing rat. Endocrinology 1999;140:844–51.ArticlePubMed
- 91. Barez-Lopez S, Montero-Pedrazuela A, Bosch-Garcia D, Venero C, Guadano-Ferraz A. Increased anxiety and fear memory in adult mice lacking type 2 deiodinase. Psychoneuroendocrinology 2017;84:51–60.ArticlePubMed
- 92. Hernandez A, Morte B, Belinchon MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by T3 in the mouse cerebral cortex. Endocrinology 2012;153:2919–28.ArticlePubMedPMC
- 93. Hernandez A, Quignodon L, Martinez ME, Flamant F, St Germain DL. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology 2010;151:5550–8.ArticlePubMedPMC
- 94. Stohn JP, Martinez ME, Zafer M, Lopez-Espindola D, Keyes LM, Hernandez A. Increased aggression and lack of maternal behavior in Dio3-deficient mice are associated with abnormalities in oxytocin and vasopressin systems. Genes Brain Behav 2018;17:23–35.ArticlePubMed
- 95. Sun Z, Xu Y. Nuclear receptor coactivators (NCOAs) and corepressors (NCORs) in the brain. Endocrinology 2020;161:bqaa083.ArticlePubMedPMC
- 96. Sakaguchi Y, Uehara T, Suzuki H, Sakamoto Y, Fujiwara M, Kosaki K, et al. Haploinsufficiency of NCOR1 associated with autism spectrum disorder, scoliosis, and abnormal palatogenesis. Am J Med Genet A 2018;176:2466–9.ArticlePubMed
- 97. Iwama K, Mizuguchi T, Takeshita E, Nakagawa E, Okazaki T, Nomura Y, et al. Genetic landscape of Rett syndrome-like phenotypes revealed by whole exome sequencing. J Med Genet 2019;56:396–407.ArticlePubMed
- 98. Sajan SA, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR, Glaze DG, et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet Med 2017;19:13–9.ArticlePubMed
- 99. Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 2016;7:13316.ArticlePubMedPMC
- 100. Yang Y, van der Klaauw AA, Zhu L, Cacciottolo TM, He Y, Stadler LK, et al. Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nat Commun 2019;10:1718.ArticlePubMedPMC
- 101. Yousefi B, Jingu H, Ohta M, Umezu M, Koibuchi N. Postnatal changes of steroid receptor coactivator-1 immunoreactivity in rat cerebellar cortex. Thyroid 2005;15:314–9.ArticlePubMed
- 102. Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet 2017;49:515–26.ArticlePubMedPMC
- 103. Vaqueiro AC, de Oliveira CP, Cordoba MS, Versiani BR, de Carvalho CX, Alves Rodrigues PG, et al. Expanding the spectrum of TBL1XR1 deletion: report of a patient with brain and cardiac malformations. Eur J Med Genet 2018;61:29–33.ArticlePubMed
- 104. Kong Y, Zhou W, Sun Z. Nuclear receptor corepressors in intellectual disability and autism. Mol Psychiatry 2020;25:2220–36.ArticlePubMedPMC
- 105. Beck-Peccoz P, Rodari G, Giavoli C, Lania A. Central hypothyroidism: a neglected thyroid disorder. Nat Rev Endocrinol 2017;13:588–98.ArticlePubMed
- 106. Bassi MT, Ramesar RS, Caciotti B, Winship IM, De Grandi A, Riboni M, et al. X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. Am J Hum Genet 1999;64:1604–16.ArticlePubMedPMC
- 107. Chung RH, Ma D, Wang K, Hedges DJ, Jaworski JM, Gilbert JR, et al. An X chromosome-wide association study in autism families identifies TBL1X as a novel autism spectrum disorder candidate gene in males. Mol Autism 2011;2:18.ArticlePubMedPMC
- 108. Qiu CH, Shimokawa N, Iwasaki T, Parhar IS, Koibuchi N. Alteration of cerebellar neurotropin messenger ribonucleic acids and the lack of thyroid hormone receptor augmentation by staggerer-type retinoic acid receptor-related orphan receptor-alpha mutation. Endocrinology 2007;148:1745–53.PubMed
- 109. Qiu CH, Miyazaki W, Iwasaki T, Londono M, Ibhazehiebo K, Shimokawa N, et al. Retinoic acid receptor-related orphan receptor alpha-enhanced thyroid hormone receptor-mediated transcription requires its ligand binding domain which is not, by itself, sufficient: possible direct interaction of two receptors. Thyroid 2009;19:893–8.ArticlePubMed
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Neuropeptides and Their Roles in the Cerebellum
Zi-Hao Li, Bin Li, Xiao-Yang Zhang, Jing-Ning Zhu
International Journal of Molecular Sciences.2024; 25(4): 2332. CrossRef - Exploring the underlying molecular mechanism of tri(1,3-dichloropropyl) phosphate-induced neurodevelopmental toxicity via thyroid hormone disruption in zebrafish by multi-omics analysis
Ying Xu, Lei Yang, Yanguo Teng, Jian Li, Na Li
Aquatic Toxicology.2023; 258: 106510. CrossRef - Association of Maternal TSH, FT4 With Children's BMI Trajectories, and Obesity: A Birth Cohort Study
Mengting Yang, Shanshan Zhang, Yuzhu Teng, Xue Ru, Linlin Zhu, Yan Han, Xingyong Tao, Hui Cao, Shuangqin Yan, Fangbiao Tao, Kun Huang
The Journal of Clinical Endocrinology & Metabolism.2023; 109(1): e190. CrossRef - Thyroid hormone receptor beta: Relevance in human health and diseases
Ghausiya Rehman, Neha Kumari, Farhad Bano, Rakesh K. Tyagi
Endocrine and Metabolic Science.2023; 13: 100144. CrossRef - Targeting Thyroid Hormone/Thyroid Hormone Receptor Axis: An Attractive Therapy Strategy in Liver Diseases
Qianyu Tang, Min Zeng, Linxi Chen, Nian Fu
Frontiers in Pharmacology.2022;[Epub] CrossRef - Histone Deacetylase 3 Inhibitor Alleviates Cerebellar Defects in Perinatal Hypothyroid Mice by Stimulating Histone Acetylation and Transcription at Thyroid Hormone-Responsive Gene Loci
Alvin Susetyo, Sumiyasu Ishii, Yuki Fujiwara, Izuki Amano, Noriyuki Koibuchi
International Journal of Molecular Sciences.2022; 23(14): 7869. CrossRef - Selection-driven adaptation to the extreme Antarctic environment in the Emperor penguin
Federica Pirri, Lino Ometto, Silvia Fuselli, Flávia A. N. Fernandes, Lorena Ancona, Nunzio Perta, Daniele Di Marino, Céline Le Bohec, Lorenzo Zane, Emiliano Trucchi
Heredity.2022; 129(6): 317. CrossRef - Long-term depression–inductive stimulation causes long-term potentiation in mouse Purkinje cells with a mutant thyroid hormone receptor
Ayane Ninomiya, Izuki Amano, Michifumi Kokubo, Yusuke Takatsuru, Sumiyasu Ishii, Hirokazu Hirai, Nobutake Hosoi, Noriyuki Koibuchi
Proceedings of the National Academy of Sciences.2022;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite