Articles
- Page Path
- HOME > Endocrinol Metab > Volume 35(4); 2020 > Article
-
Original ArticleClinical Study Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population
-
Heewon Choi1
 , Kyoung Jin Kim2,3, Namki Hong2, Saeam Shin4, Jong-Rak Choi4, Sang Wook Kang5, Seung Tae Lee4, Yumie Rhee2
, Kyoung Jin Kim2,3, Namki Hong2, Saeam Shin4, Jong-Rak Choi4, Sang Wook Kang5, Seung Tae Lee4, Yumie Rhee2 -
Endocrinology and Metabolism 2020;35(4):858-872.
DOI: https://doi.org/10.3803/EnM.2020.683
Published online: December 23, 2020
1Yonsei University College of Medicine, Seoul, Korea
2Department of Internal Medicine, Severance Hospital, Endocrine Research Institute, Yonsei University College of Medicine, Seoul, Korea
3Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea
4Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Korea
5Thyroid-Endocrine Surgery Division, Department of Surgery, Yonsei University College of Medicine, Seoul, Korea
- Corresponding authors: Seung Tae Lee. Department of Laboratory Medicine, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea, Tel: +82-2-2228-2450, Fax: +82-2-313-0956, E-mail: lee.st@yuhs.ac
- Yumie Rhee. Department of Internal Medicine, Severance Hospital, Endocrine Research Institute, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea, Tel: +82-2-2228-1973, Fax: +82-2-393-6884, E-mail: yumie@yuhs.ac
Copyright © 2020 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Pheochromocytoma and paragangliomas (PPGL) are hereditary in approximately 30% to 40% cases. With the advancement of genetic analysis techniques, including next-generation sequencing (NGS), there were attempts to classify PPGL into molecular clusters. With NGS being applied to clinical settings recently, we aimed to review the results of genetic analysis, including NGS, and investigate the association with clinical characteristics in Korean PPGL patients.
-
Methods
- We reviewed the medical records of PPGL patients who visited Severance hospital from 2006 to 2019. We documented the clinical phenotype of those who underwent targeted NGS or had known germline mutations of related genes.
-
Results
- Among 57 PPGL patients, we found 28 pathogenic germline mutations of susceptibility genes. Before the targeted NGS was implemented, only obvious syndromic feature lead to the Sanger sequencing for the specific genes. Therefore, for the exact prevalence, only patients after the year 2017, when targeted NGS was added, were included (n=43). The positive germline mutations were found in 14 patients; thus, the incidence rate is 32.6%. Patients with germline mutations had a higher likelihood of family history. There were significant differences in the type of PPGLs, percentage of family history, metastasis rate, presence of other tumors, and biochemical profile among three molecular clusters: pseudohypoxic tricarboxylic acid cycle-related, pseudohypoxic von Hippel-Lindau (VHL)/endothelial PAS domain-containing protein 1-related, and kinase-signaling group. Germline mutations were identified in seven PPGL-related genes (SDHB, RET, VHL, NF1, MAX, SDHA, and SDHD).
-
Conclusion
- We report the expected prevalence of germline mutations in Korean PPGL patients. NGS is a useful and accessible tool for genetic analysis in patients with PPGLs, and further research on molecular classification is needed for precise management.
- Pheochromocytoma (PCC) and paraganglioma (PGL) are tumors originating from chromaffin cells in the adrenal medulla and extra-adrenal paraganglia of the sympathetic and parasympathetic nervous chains, respectively [1]. These neuroendocrine tumors are collectively referred to as pheochromocytoma and paraganglioma (PPGL). PCCs and sympathetic PGLs can synthesize and secrete catecholamines, mainly adrenaline and noradrenaline, and even dopamine, as they originate from neuroendocrine cells derived from the embryonic neural crest, while parasympathetic PGLs rarely produce catecholamines [2,3].
- PPGLs are rare tumors with a incidence of 0.95 per 100,000 person-years in the United States. They account for 0.02% to 0.5% of patients with hypertension [4]. Classically, PCC was called “10% tumor,” as this tumor had a 10% rate of malignancy, bilateral occurrence, pediatric-onset, being asymptomatic, familial inheritance, and having an extra-adrenal location [5,6]. Regarding the pathogenesis, 90% of PCCs were considered sporadic, with only 10% being hereditary with germline mutations [7]. However, this rule was disproved as techniques for genetic testing evolved, and the prevalence of PPGL with germline mutations was reported to be approximately 30% to 40% [8–10].
- A traditional familial cause of PPGL was noted in syndromes such as multiple endocrine neoplasia (MEN) type IIA and IIB, neurofibromatosis (NF) type 1, and von Hippel-Lindau (VHL) disease [5]. However, with the advancement of genetic analysis techniques, more susceptibility genes for hereditary PPGL syndromes have been discovered. Mutations in subunits of the succinate dehydrogenase (SDH) complex, a mitochondrial enzyme for oxidative phosphorylation [11], is now a well-known cause of hereditary PPGL [12,13]. An astonishing feature of hereditary PPGLs is the associated high degree of genetic heterogeneity. Some of the susceptibility or driver genes, mutations known to cause hereditary PPGLs [9], are involved in the hypoxic response during cellular metabolism. Others are involved in kinase signaling pathways [14]. Due to the complexity of analyzing the genetics of PPGLs, there has been an attempt to approach this issue by classifying the genes into molecular clusters: pseudohypoxia, Wnt signaling, and kinase signaling group [15].
- The high prevalence of PPGLs is known to be caused by a certain germline mutation of susceptibility genes, making them the most heritable tumor in humans [9]. Therefore, genetic testing is recommended in all patients regardless of family history [16]. Genetic testing has become more accessible with the utilization of next-generation sequencing (NGS), an accurate, fast, and inexpensive way of genetic testing and has shifted the paradigm of genetic research. The NGS in PPGL (NGSnPPGL) study group suggested that targeted NGS is the most preferred method for genetic testing of PPGL patients [17]. Targeted NGS analyzes only the genes in the panel previously reported to have an association with tumorigenesis of PPGLs.
- Regarding the development of valuable NGS technology, more susceptibility genes for hereditary PPGLs are being discovered, but the relative frequencies of the mutations found to vary between studies and geographic locations [17–19]. Moreover, since PPGL is a very rare type of tumor, large-scale studies on clinical relationships are limited. The genotype-phenotype relationship has not been thoroughly examined, especially in Korea. Regarding the clinical features of these tumors, a study by Kim et al. [20] on 119 patients with PCCs from 1986 to 2009 is so far the largest in Korea; however, it only included PCCs and not PGLs. Further, this study lacked information on the genetic aspect of this issue. Another study by Kim et al. [21] demonstrated a correlation between germline mutations and the clinical characteristics of PPGLs by examining 53 patients with non-syndromic PPGLs. In this study, seven patients (13.2%) had germline mutations: rearranged during transfection (RET; n=2), VHL (n=1), SDHB (n=2), and SDHD (n=2). The prevalence rate of germline mutations is much lower than the recently reported prevalence worldwide. This is due the fact that the information on germline mutations in this study was limited to only four genes screened by direct sequencing. Other publications on this topic are limited to case reports. Since NGS has been applied to the clinical hospital setting only recently in Korea, there is no report examining germline mutations of susceptibility genes related to PPGLs in Korea.
- We aimed to investigate the prevalence of gene mutations found in PPGL patients in the Korean population and to examine its associated clinical characteristics using genetic analyses, including targeted NGS. To this end, we reviewed the clinical and genetic features of patients with PPGLs in Severance Hospital.
INTRODUCTION
- Patients
- We reviewed the medical records of 78 patients with the histopathological or clinical diagnosis of PPGLs who visited Severance Hospital, Seoul, Korea between 2006 and 2019, and have been managed by a single, well-experienced endocrinology specialist (corresponding author Y.R.). Histopathological diagnosis of PPGL was based on histologic confirmation by pathologists. Four patients did not undergo surgery due to having an unresectable mass or individual preference. However, they were clinically diagnosed with PCCs because of the apparent adrenal mass on imaging studies such as computed tomography (CT), positron emission tomography, or iodine-131-labeled metaiodobenzylguanidine scintigraphy and elevated catecholamine level in the plasma or urine.
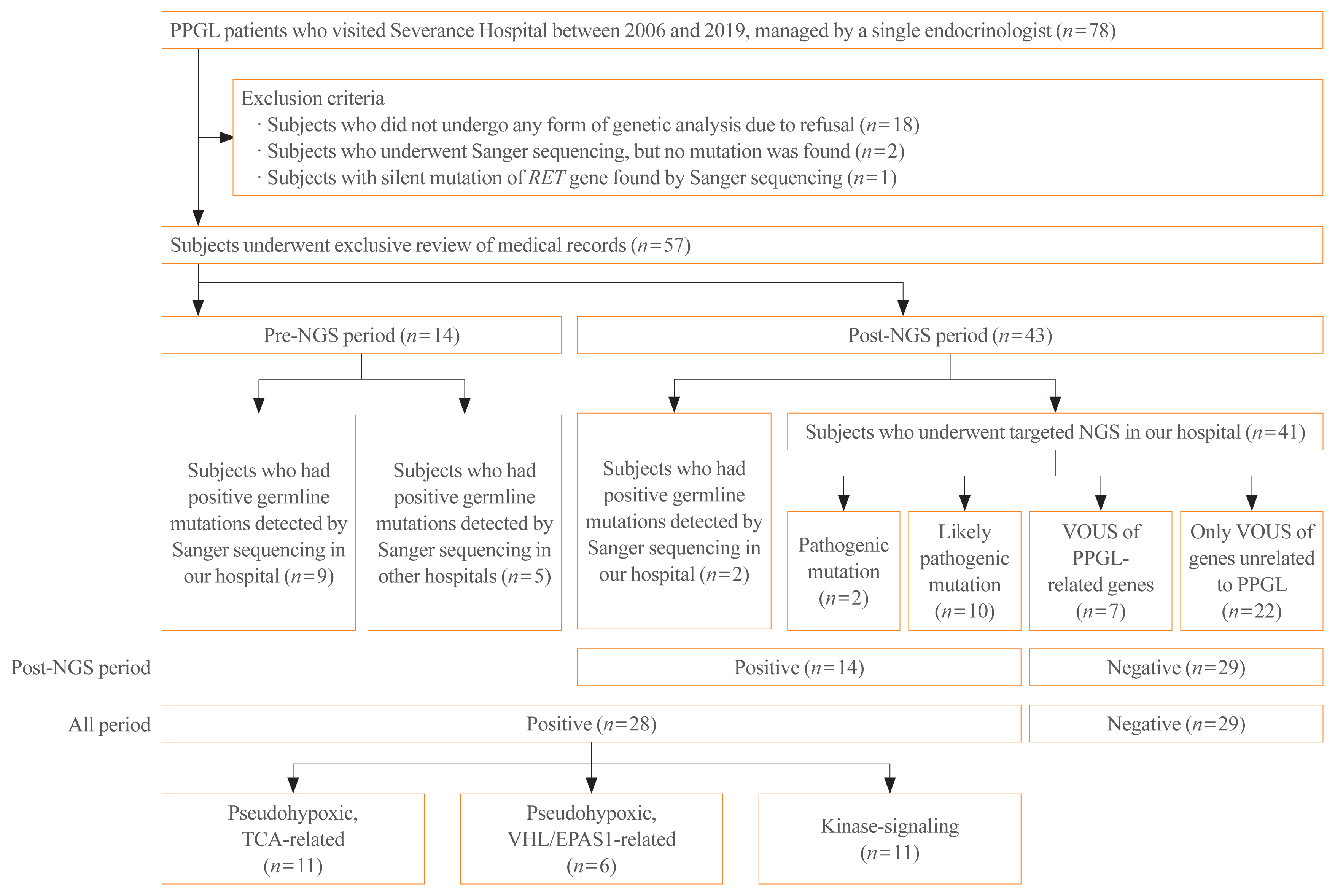
- Of the 78 patients, 57 were selected for inclusion in this study (Fig. 1). Patients who refused to undergo any form of genetic testing due to personal or economic reasons were excluded (n=18). Also, patients who underwent Sanger sequencing, but no mutation was found, were excluded (n=2). These exclusion criteria were applied since we could not evaluate the genetic background or rule out the possibility of patients having other meaningful mutations in terms of the pathogenesis of PPGLs, since the Sanger sequencing included only a very limited number of genes (i.e., RET, NF1, VHL). In the same context, a patient who had a silent mutation in the RET gene by Sanger sequencing was also excluded (n=1) because it was considered as having no mutation. As a result, the included patients are those who underwent targeted NGS in our hospital (n=41), and those who had germline mutations found either by Sanger sequencing in our hospital (n=11) or in other hospitals (n=5).
- The medical records of the 57 patients were thoroughly reviewed. Information, including age, age of onset, sex, family history, location of the tumor, tumor size, metastasis, recurrence, presence of other tumors, discovery route, symptoms and signs, histologic examination results, and biochemical profile, were extracted. When a patient had at least one family member with PPGL or second-degree relatives with a syndrome related to PPGL, the patient was considered to have a family history. The tumor size was represented by its largest diameter measured histologically or on images if histological findings were not present. Only cases with distant metastasis counted as malignant PPGL, conservatively with high specificity but relatively low sensitivity [22]. It is known that distant metastasis is the only reliable factor that can distinguish malignant PPGL among other clinical, biochemical, or histological features [23,24]. The time at which malignancy was detected was also recorded. Recurrence was defined as the reappearance of PPGL, i.e., presence of a newly discovered mass on imaging studies with elevation in catecholamines levels after the complete removal of the tumor by surgical resection [25,26]. Recurrence included both local and distant recurrence. The tumor discovery route was classified as an incidental finding on CT or ultrasonography regardless of the presence of symptoms and suspected tumors based on the presenting symptoms known to correlate with PPGL when the patient visited the hospital. An incidental finding of a mass in the adrenal gland is called adrenal incidentaloma, and additional workup was done for further evaluation and proper diagnosis of the mass. Uncommon discovery routes included the detection of gene mutations before PCC was detected in a family study. For biochemical profile, plasma or 24-hour urine epinephrine, metanephrine, norepinephrine, normetaepinephrine, dopamine, and urinary vanillylmandelic acid levels were measured. PPGL was considered functional if the catecholamine levels were elevated beyond the normal upper limit, adrenergic when epinephrine/metanephrine level was predominantly elevated; noradrenergic when norepinephrine/normetanephrine level was predominantly elevated; and adrenergic/noradrenergic when both epinephrine/metanephrine and norepinephrine/normetanephrine levels were elevated. When all catecholamine levels were normal, the tumor was classified as silent [27].
- The research protocol was approved by the Institutional Review Board of Severance Hospital, South Korea (IRB No. 4-2019-4088). Informed consent was waived by the board.
- Genetic analyses
- Targeted NGS was strongly recommended to all patients who were diagnosed with PPGLs as a routine test since January 2017. Before targeted NGS was implemented in our hospital (pre-NGS period), Sanger sequencing was conducted selectively in patients who demonstrated features of familial PPGLs, such as MEN2, NF1, and VHL disease. Even after adopting targeted NGS (post-NGS period), Sanger sequencing was the diagnosis of choice for patients who had syndromic features that strongly implied specific gene mutations and family members of patients with germline mutations.
- We conducted targeted NGS in 41 patients with PPGLs from March 2017 to October 2019. We used our hospital’s novel NGS panel to detect gene mutations of 400 genes for hereditary endocrine disorders (Supplemental Table S1). There were 19 PPGL-related genes in the panel (alpha thalassemia mental retardation syndrome X-linked [ATRX], BRAF, Egl-9 family hypoxia-inducible factor 1 [EGLN1], endothelial PAS domain-containing protein 1 [EPAS1], fibroblast growth factor receptor 1 [FGFR1], fumarate hydratase [FH], Harvey rat sarcoma viral oncogene homolog [HRAS], kinesin family member 1B [KIF1B], Myc-associated protein X [MAX], NF1, RET, SDH A/AF2/B/C/D [SDHA/SDHAF2/SDHB/SDHC/SDHD], transmembrane protein 127 [TMEM127], tumor protein 53 [TP53], and VHL) [17]. Our hospital’s targeted NGS followed the 2017 American College of Medical Genetics and Genomics (ACMG) guidelines [28]. Of the five categories in ACMG guidelines, our hospital only reported three categories: pathogenic, likely pathogenic, and variant of unknown significance (VOUS).
- When a pathogenic or likely pathogenic mutation was found by targeted NGS or germline mutation of PPGL-related susceptibility gene was detected by Sanger sequencing, a patient was considered to have mutation of known pathogenicity. If a patient had a mutation of known pathogenicity, the genetic analysis result counted as positive, which indicates PPGL with a germline mutation of a susceptibility gene. If a patient had VOUS mutations only, the genetic analysis result was considered negative. Meanwhile, some patients had VOUS mutations of genes that are known to be related to the pathogenesis of PPGL. Although physician assumed that these VOUS of PPGL-related genes might have clinical significance, they were not considered positive as the pathogenicity of mutations has not been proven yet due to insufficient data.
- For patients with germline mutations of PPGL-related genes, information on mutated genes and specific loci were collected. Mutated genes were classified into molecular clusters as previously described: pseudohypoxia, Wnt signaling, and kinase signaling group [15].
- Family study
- Family members of genetically confirmed patients were recommended to undergo genetic analysis. Fourteen individuals, who were family members of nine PPGL patients with germline mutations, underwent Sanger sequencing for the same mutation as detected in the respective family member with PPGL.
- Statistical analysis
- Clinical characteristics were presented as mean±standard deviation for parametric continuous variables, as median (interquartile range) for nonparametric continuous variables, and as counts (percentage) for categorical variables. For continuous variables, the Shapiro-Wilk test was used to test for normality. Clinical characteristics were compared according to the genetic analysis results and molecular clusters of patients of all periods. For parametric continuous variables, comparisons were made using a t test between two groups and one-way analysis of variance (ANOVA) between three groups. Comparisons were conducted using the Kruskal-Wallis test for nonparametric continuous variables and chi-square test for categorical variables. A two-sided P<0.05 was considered to be statistically significant. All statistical analyses were performed using Stata version 14.2 (Stata Corp., College Station, TX, USA).
METHODS
- Patient characteristics
- Of the 78 patients with PPGLs, 61 patients (78.2%) were diagnosed with PCC, and the other 17 patients (21.8%) were diagnosed with PGL. After excluding patients, we extensively reviewed medical records of 57 patients (73.1%) who underwent targeted NGS or were found to have germline mutations of a susceptibility gene by direct sequencing. The descriptive clinical characteristics of these patients are presented in Table 1. The mean age of onset was 42.8±14.8 years, and 31 patients (54.4%) were females. Forty patients (70.2%) were diagnosed with PCC, and the other 17 (29.8%) were diagnosed with PGL.
- The location of PCC was either the right (40.0%), left (40.0%), or bilateral (20.0%) adrenal glands. While three PGLs (17.7%) were parasympathetic as they were located in the head and neck region, including the mediastinum, 14 PGLs (82.4%) were sympathetic, located in thoracoabdominopelvic regions, including retroperitoneum, pancreas, para-aortic space, and bladder [29]. All of parasympathetic PGLs were biochemically silent. The median tumor size was 3.7 cm. The metastasis and recurrence rates were 26.3% and 19.3% for PPGL, respectively. Among 15 patients with metastasis, seven cases were synchronous, whereas eight cases were metachronous. The site of distant metastasis included bone (n=5), lymph nodes (n=5), liver (n=3), and lungs (n=3). Eighteen patients (31.6%) had previously diagnosed tumors in other organs: medullary thyroid carcinoma (MTC, n=8), papillary thyroid carcinoma (n=3), renal cell carcinoma (n=3), and hemangioblastoma (n=3) among others. Forty-three patients presented with symptoms related to PPGLs, including hypertension (n=26), palpitation (n=17), headache (n=8), weight loss (n=6), abdominal pain (n=5), dizziness (n=4), and sweating (n=3). PPGL was detected incidentally by CT or ultrasonography in 21 patients (41.2%), whereas 28 patients (54.9%) had symptoms of PPGL at the first visit. In two patients (3.9%), gene mutations were first detected during a family study, and the following workup revealed the presence of PCCs. Functional tumors accounted for 70.2% of the cases, with the noradrenergic type being the most common (52.6%).
- Germline mutations in PPGL patients
- Fourteen patients received Sanger sequencing in pre-NGS period, while the other 43 patients underwent genetic testing by either direct sequencing (n=2) or targeted NGS (n=41) in post-NGS period.
- Among patients of all periods, 28 patients had positive genetic analysis results, as they were found with germline mutations of PPGL-related genes with pathogenicity, detected either by targeted NGS (n=2 for pathogenic, n=10 for likely pathogenic) or Sanger sequencing (n=16). The other 29 patients were regarded to have negative genetic analysis results. Among those with negative results, seven patients had VOUS mutations of susceptibility genes, while 22 patients had only VOUS mutations of genes unrelated to PPGL.
- Meanwhile, to obtain a more accurate prevalence of germline mutations among PPGL patients, we took into consideration only the patients who received gene analyses during post-NGS period. Since 2017, the year at which targeted NGS was recommended to all patients, a total of 43 PPGL patients underwent genetic testing (n=41 for targeted NGS, n=2 for Sanger sequencing). Among those 43 patients, 14 patients had positive results, so the proportion of germline mutations is estimated to be 32.6% (14 out of 43).
- Comparison of clinical characteristics, including age, age of onset, sex, family history, location of the tumor, tumor size, metastasis, time of metastasis discovery, recurrence, presence of other tumors, discovery route, and biochemical profile, between positive and negative genetic analysis results is presented in Table 1. PPGL patients with germline mutations were more likely to have a family history (P=0.001). Although not statistically significant, patients with germline mutations tended to have a lower age of onset and prevalence of bilateral PCCs and a higher rate of metastasis and recurrence.
- Among 28 patients with positive results, 19 different germline mutations from seven susceptibility genes were discovered. The most frequently mutated genes were SDHB (n=9), followed by RET (n=8), VHL (n=6), NF1 (n=2), MAX (n=1), SDHA (n=1), and SDHD (n=1) (Fig. 2). Sixteen patients had classical familial syndromes. Of these, eight had MEN2, six had VHL syndrome, and two had NF type 1. Meanwhile, seven VOUS mutations were found in four types of genes related to PPGL; SDHB (n=2), EPAS2 (n=2), KIF1B (n=1), SDHC (n=1), and TMEM127 (n=1) (Supplemental Table S2).
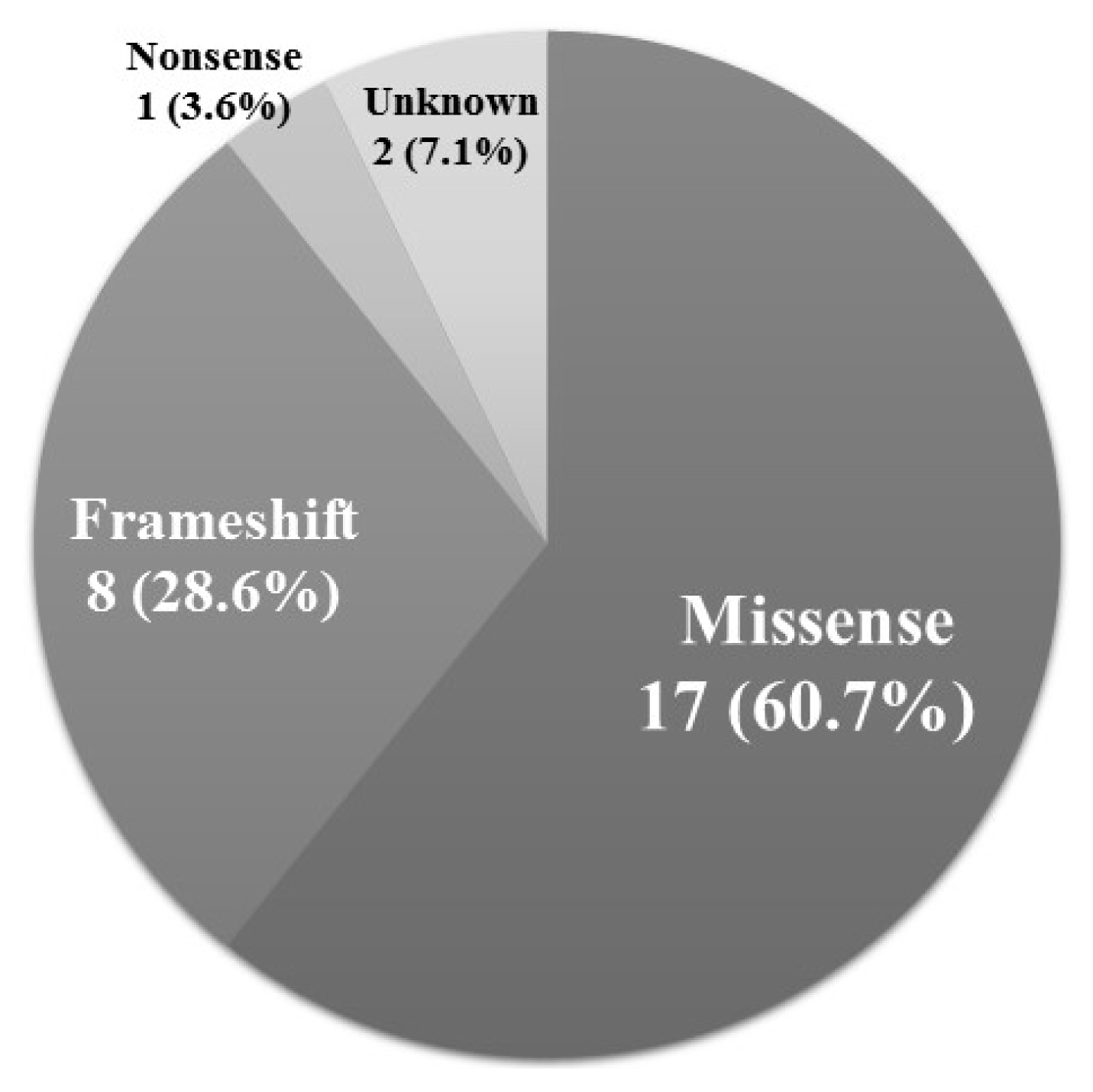
- Seventeen out of the 28 positive mutations (60.7%) were missense mutations (Fig. 3). Eight mutations (28.6%) were frameshift mutations, and one mutation (3.6%) was a nonsense mutation.
- Mutated genes with pathogenicity were classified into three molecular clusters: pseudohypoxic tricarboxylic acid (TCA) cycle-related group including SDHB, SDHA, and SDHD (n=11); pseudohypoxic VHL/EPAS1-related group including VHL (n=6); and a kinase-signaling group including RET, NF1, and MAX (n=11). Clinical characteristics were compared between the molecular clusters (Table 2). The diagnosis of PCC or PGL significantly differed between the clusters (P=0.003). While the TCA cycle-related group patients were comparable in terms of the type of PPGL (PCC 36.4% vs. PGL 63.6%), the kinase signaling group consisted of PCCs only (PCC 100.0% vs. PGL 0.0%). Patients in the kinase signaling group were more likely to have a family history (63.6%), followed by those in the pseudohypoxic VHL/EPAS1-related group (33.3%), while the pseudohypoxic TCA-cycle related group had no family history (P= 0.006). Metastasis rate was the highest in the pseudohypoxic TCA cycle-related group (63.6%), followed by the pseudohypoxic VHL/EPAS1-related group (50.0%), and the kinase signaling group had no patient with distant metastasis (P=0.006). The kinase signaling group had the highest percentage of patients who had other tumors (72.7%), followed by the pseudohypoxic VHL/EPAS1-related (50.0%) and pseudohypoxic TCA cycle-related (9.1%) groups (P=0.010). A significant difference in biochemical profile between the molecular clusters was detected (P=0.036). Notably, functional PPGLs in the pseudohypoxic group were all noradrenergic type. Although there was no statistically significant difference, all the pseudohypoxic TCA cycle-related group cases had unilateral PCC. Head and neck PGL was detected in only the pseudohypoxic TCA cycle-related group.
- Detailed clinical and genetic characteristics, including the specific mutation loci, of patients with positive genetic analyses results are presented in Table 3. Information on patients with VOUS of PPGL-related genes can be found at Supplemental Table S2. While most of the genes have already been reported in previous studies, four novel mutations were discovered in the present study: EPAS1 c.1250G>A (p.Gly417Glu), NF1 c.6215delA (p.His2072LeufsTer10), NF1 c.6777del (p.Gly2260fs), and SDHC exon 2–6 duplication.
- Family study
- Fourteen family members of nine patients underwent Sanger sequencing for family study. Eight of these family members (57.1%) had the same germline mutation as that of the patient: SDHB (n=4), RET (n=3), and NF1 (n=1). Those with germline mutations underwent screening workup, including history taking, physical examination, measurement of plasma or urine catecholamine levels, and imaging such as CT for the detection of PPGLs. For genes known to cause syndromic diseases, further evaluation was performed, i.e., thyroid sonography for patients with RET mutation considering the possibility of MTC. As a result, two persons were newly diagnosed with PCCs. Both daughters of a patient with PCC and MEN2A (Case no. 17) had the same RET gene mutation. While one only had C-cell hyperplasia, another (Case no. 13) was diagnosed with PCC, MTC, and MEN2A. Similarly, the daughter (Case no. 18) of a patient with PCC and MEN2A (Case no. 20) was found to have RET gene mutation and was diagnosed with PCC, MTC, and MEN2A. The daughter of another patient with PCC and NF1 (Case no. 12) was also diagnosed with NF1 due to the same mutation in the NF1 gene; although she did not have PPGL, typical clinical features of neurofibromatosis, café-au-lait spots, and neurofibroma were noted. Four family members who had SDHB mutations currently showed no phenotype, as no evidence of PPGL was found after thorough screening workup, including biochemical and radiological tests, and they did not have any symptoms and signs. This is probably due to the low penetrance of the SDHB gene [30] and the possibility of delayed manifestation.
RESULTS
- Of the 78 Korean patients with PPGLs, we exclusively evaluated 57 patients and examined the clinical and genetic features of the disease. Among those 57 patients who underwent either targeted NGS or Sanger sequencing, 28 patients were found to have pathogenic germline mutations of PPGL-related genes. Considering the fact that targeted NGS was implemented recently as a routine modality, prevalence rate of germline mutation is 32.6% (14 out of 43). There was a higher likelihood of family history of PPGL in patients with germline mutations than those with no mutations. There were significant differences in the type of PPGL, percentage of family history, metastasis rate, the presence of other tumors, and biochemical profile between three molecular clusters. Nineteen different germline mutations from seven genes (SDHB, RET, VHL, NF1, MAX, SDHA, and SDHD) were found, most of which were missense mutations. Seven patients had VOUS mutations of PPGL-related genes (SDHB, EPAS1, KIF1B, and SDHC). EPAS1, KIF1B, MAX, SDHA, SDHC, and TMEM127 mutations were the first to be reported among Korean patients with PPGL.
- In this study, 70.2% of patients were diagnosed with PCCs, while 29.8% were diagnosed with PGLs, which was in line with the previously reported percentage of PCC (69% to 89%) [18,22,31]. The mean age of onset in this study was 42.8 years, and 54.4% of the patients were women. This information is concordant with that reported in a previous study (the peak age of onset, 40 to 50 years). Furthermore, PPGLs were reported to have an approximately equal distribution among men and women [31]. Family history was found only in nine patients (15.5%), all of whom were found with germline mutations. However, most of the patients with germline mutations did not have family histories. A previous study also found that a high incidence of PCCs positive for mutations occurred without a family history [32]. They suggested a potential explanation for this phenomenon: a high rate of spontaneous mutations, low penetrance of the gene mutations, or maternal imprinting.
- Malignant PPGL, defined as the presence of distant metastasis, is reported to have a prevalence of approximately 10% [16, 22,24]. However, in our data, the metastatic rate was as high as 26.3% because our center is a tertiary referral hospital. Frequent sites of metastases were the bone, lymph nodes, liver, and lung, which were concordant with a previous study [33]. It is known that the risk of metastasis is higher in extra-adrenal location, with a higher level of plasma methoxytyramine (metabolites of dopamine), and in SDHB mutations [34–36]. Our study only showed a significant difference in metastatic rate depending on SDHB mutations. Among 15 patients with metastatic PPGL, six patients had SDHB mutations, three patients had VHL mutations, one patient had SDHA mutation, and the other five patients were sporadic.
- A previous systematic review had shown that the incidence of recurrence, including local and systemic recurrence, and appearance of new tumors, is 0.95 per 100 person-years, which is 4.7% of occurrence in 5 years [22]. One-fifth of the patients were found to have the recurrent disease in this study, similar to the previous report [20]. However, some studies have reported that the presence of syndromic disease, larger tumor size, and PGL relative to PCC being related to an increased risk of recurrence [22]; our study found no association between them. Owing to varying results between studies and the absence of precise features that predict recurrence, all patients should undergo follow-up, including clinical history-taking, imaging, and biochemical evaluation.
- With the development of imaging techniques, the rate of incidental diagnosis of PCC dramatically increased to nearly 30% [37]. In Korea, this rate was even higher, with 36.1% reported in a previous study [20]. CT was the frequently used diagnostic tool because the cost is lower than that in other countries due to the National Health Insurance System. In our study, PPGLs were discovered incidentally in 41.2% of the cases, showing that the rate of incidental diagnosis is steadily increasing. This phenomenon could be attributed to the growing accessibility of imaging procedures, including abdominal CT and sonography. Meanwhile, in 54.9% of the cases, the tumor was discovered because of the symptoms and signs. Similar to that in a previous study, hypertension was the most common clinical sign, whereas palpitation and headache were the most common symptoms [7]. About half had either paroxysmal or sustained hypertension, and three-fourth of the patients had symptoms and signs associated with PPGLs. Among functional PPGLs, the noradrenergic type was the most common. There was no significant difference in the biochemical profile between PPGLs with and without germline mutations.
- To our knowledge, the current result on the frequency of germline mutations in Korean PPGL patients, 32.6%, is relatively higher than the previously reported prevalence in Korean so far (13.2%). However, this is concordant with the worldwide prevalence of germline mutations in PPGL known to be around 30% to 40% [8–10]. Similar to previous studies, SDHB mutation was the most common [17,38]. VOUS mutations were excluded from significant germline mutations due to lack of data to support their pathogenicity. Indeed, VOUS mutations found in our study were from SDHB, EPAS1, SDHC, and TMEM127 genes, and except for SDHB, other genes are known to be extremely rare in frequencies [17,38]. Nevertheless, as VOUS indicates “missing pathogenicity,” the possibility of reclassification to other ACMG classes exists with the additional accumulation of clinical data [39].
- We also investigated the clinical characteristics based on the molecular clusters. Genes were classified into three molecular clusters, pseudohypoxic TCA cycle-related (known to be 10% to 15% of PPGLs), pseudohypoxic VHL/EPAS1-related (15% to 20%), and the kinase signaling (50% to 60%) group. In our study, there were significant differences in the type of PPGLs, percentage of family history, metastasis rate, the presence of other tumors, and biochemical profile. While PPGLs related to the TCA cycle can occur in any location, PPGLs related to VHL/EPAS1 are not likely to be located on the head and neck areas. Kinase signaling-related PPGLs are most likely to be PCCs with an uncommon incidence of parasympathetic PGLs, and sympathetic PGLs are not likely to be present [15]. These findings are concordant with our data that the pseudohypoxic TCA cycle-related group included all of PCCs, sympathetic and parasympathetic PGLs, but the pseudohypoxic VHL/EPAS1-related group only included PCCs and parasympathetic PGLs. Most of the patients in the kinase signaling group had PCCs except one with parasympathetic PGL. Our result showed that the kinase signaling group had 50% of family history but none in the pseudohypoxic TCA cycle-related group. The discrepancies between the groups must be attributed to the rate of spontaneous mutation and penetrance. A previous review stated that the risk of metastasis is highest in the pseudohypoxic TCA cycle-related group and lowest in the kinase signaling group, and the result was largely the same in our data. Pseudohypoxic TCA cycle-related PPGLs are reported to have the lowest paraganglia cell differentiation with a decreased expression of catecholamine-synthesizing enzymes leading to depressed catecholamine levels. Pseudohypoxic VHL/EPAS1-related PPGLs have an intermediate level of catecholamine production, and kinase-signaling PPGLs, being the most differentiated, are likely to be most functional [15]. Our study results are in line with those of a previous study, which reported that pseudohypoxic PPGLs are purely noradrenergic and never adrenergic because they always lack phenylethanolamine N-methyltransferase, an enzyme that catalyzes the synthesis of epinephrine from norepinephrine [15].
- There are several limitations to this study. First, our study population has a small sample size in a single center, so our results might have restricted value in terms of generalization. However, considering the rare prevalence of PPGL, an analysis of 57 patients of clinical and genetic characteristics in Korea is noteworthy. Second, this study has a potential for biases. We included PPGL patients managed by a single endocrinologist. Although vast majority of PPGL patients in our hospital were treated by this physician, few patients might have been missed by being registered to other endocrinologists or endocrine surgeons. Also, we excluded two patients with no mutations and one patient with silent mutation analyzed by only Sanger sequencing but included 17 patients in whom germline mutations were found. This exclusion was inevitable due to the characteristic of Sanger sequencing, by which we can only seek for mutation at a gene of interest, but this could have created selection bias. To minimize the bias, the frequency of germline mutation was calculated among the patients who underwent genetic analyses after the targeted NGS technique was adopted as a routine test. Consecutive use of targeted NGS by recommending it routinely to all the PPGL patients will be necessary for future studies. Difficult interpretation of VOUS resulted from targeted NGS was one of the greatest problems in our study. We approached this issue in rather conservative manner, as we considered VOUS as negative. This might have led to underestimation of the frequency as we acknowledge the possibility that some VOUS mutations might be reclassified to pathogenic or likely pathogenic over time with additional reports of data. Meanwhile, patients who had no mutations of susceptibility genes by targeted NGS cannot be considered as patients with absolutely negative results, as we only analyzed the known susceptibility genes of PPGLs. For those patients with negative results with NGS, we might recommend whole-genome sequencing [17]. Whole-exome sequencing of these patients might uncover new susceptibility genes that are still unknown, but the validation of the causative relationship between the mutation and the PPGL will be a difficult process. Moreover, epigenetic changes were not taken into consideration in this study. Previous studies have shown that epigenetic gene silencing of tumor suppressor genes due to promotor methylation is present and considered pathogenic in some patients with PPGLs [40,41]. This finding suggests that the percentage of PPGL patients with genetic background might be higher than the value presented.
- In conclusion, our data present the frequency of germline mutations in Korean patients with PPGL. The expected prevalence of germline mutations of susceptibility genes in PPGL patients is estimated to be approximately 32.6%. With the utilization of NGS, genetic testing has become more accessible, simultaneously providing us with abundant information. Further research into the molecular characterization of PPGL can lead us to pursue and take a step toward the concept of precision medicine.
DISCUSSION
SUPPLEMENTARY MATERIAL
Supplemental Table S1.
Supplemental Table S2.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: Y.R. Acquisition, analysis, or interpretation of data: H.C., N.H., S.S., J.R.C., S.W.K., S.T.L. Drafting the work or revising: H.C., K.J.K. Final approval of the manuscript: Y.R.
Article information
-
Acknowledgements
- This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea. (grant number: HI19C0189)

| Characteristic | Total (n=57) | Genetic analysis result | P valuea | |||
|---|---|---|---|---|---|---|
|
|
||||||
| Positive (n=28) | Negative (n=29) | |||||
|
|
||||||
| All period (n=28) | Pre-NGS period (n=14) | Post-NGS period (n=14) | ||||
| Age, yrb | 48.5±13.7 | 46.2±15.0 | 48.1±15.7 | 48.5±13.7 | 50.8±12.2 | 0.211 |
|
|
||||||
| Age of onset, yrb | 42.8±14.8 | 39.5±15.9 | 42.2±18.2 | 42.8±14.8 | 46.0±13.1 | 0.096 |
|
|
||||||
| Female sex | 31 (54.4) | 16 (57.1) | 7 (50.0) | 9 (64.3) | 15 (51.7) | 0.681 |
|
|
||||||
| Family history | 9 (15.8) | 9 (32.1) | 2 (14.3) | 7 (50.0) | 0 | 0.001 |
|
|
||||||
| Type of PPGL | ||||||
| PCC | 40 (70.2) | 20 (71.4) | 7 (50.0) | 13 (92.9) | 20 (71.4) | 0.839 |
| PGL | 17 (29.8) | 8 (28.6) | 7 (50.0) | 1 (7.1) | 8 (28.6) | |
|
|
||||||
| Location | ||||||
| PCC (n=40) | ||||||
| Unilateral adrenal gland | 32 (80.0) | 14 (70.0) | 4 (57.1) | 10 (76.9) | 18 (90.0) | 0.114 |
| Right adrenal gland | 16 (40.0) | 8 (40.0) | 2 (28.6) | 6 (46.2) | 8 (40.0) | |
| Left adrenal gland | 16 (40.0) | 6 (30.0) | 2 (28.6) | 4 (30.8) | 10 (50.0) | |
| Bilateral adrenal glands | 8 (20.0) | 6 (30.0) | 3 (42.9) | 3 (23.1) | 2 (10.0) | |
| PGL (n=17) | ||||||
| Head and neck | 3 (17.7) | 1 (12.5) | 1 (14.3) | 0 | 2 (22.2) | 0.600 |
| Othersc | 14 (82.4) | 7 (87.5) | 6 (85.7) | 1 (100.0) | 7 (77.8) | |
|
|
||||||
| Tumor size, cm (n=48)d | 3.7 (2.7–4.8) | 3.5 (2.0–4.6) | 3.1 (2.0–4.4) | 4.1 (3.2–4.7) | 3.8 (3.2–5.0) | 0.261 |
|
|
||||||
| Metastasis | 15 (26.3) | 10 (35.7) | 6 (42.9) | 4 (28.6) | 5 (17.2) | 0.113 |
| Synchronous | 7 (12.3) | 4 (14.3) | 3 (21.4) | 3 (21.4) | 3 (10.3) | 0.464 |
| Metachronous | 8 (14.0) | 6 (21.4) | 3 (21.4) | 1 (7.2) | 2 (6.9) | |
|
|
||||||
| Recurrence | 11 (19.3) | 7 (25.0) | 4 (28.6) | 3 (21.4) | 4 (13.8) | 0.284 |
|
|
||||||
| Presence of other tumors | 18 (31.6) | 12 (42.9) | 3 (21.4) | 9 (64.3) | 6 (20.1) | 0.072 |
|
|
||||||
| Discovery route (n=51) | ||||||
| Incidental | 21 (41.2) | 11 (45.8) | 4 (33.3) | 7 (58.3) | 11 (37.0) | 0.224 |
| Symptomatic | 28 (54.9) | 11 (45.8) | 7 (58.3) | 4 (33.3) | 17 (63.0) | |
| Gene mutation | 2 (3.9) | 2 (8.3) | 1 (8.3) | 1 (8.3) | 0 | |
|
|
||||||
| Biochemical profile | ||||||
| Adrenergic/Noradrenergic | 9 (15.8) | 4 (14.3) | 1 (7.1) | 3 (21.4) | 5 (17.2) | 0.732 |
| Adrenergic | 1 (1.8) | 1 (3.6) | 0 | 1 (7.1) | 0 | |
| Noradrenergic | 30 (52.6) | 14 (50.0) | 9 (64.3) | 5 (35.7) | 16 (55.2) | |
| Silent | 17 (29.8) | 9 (32.1) | 4 (28.6) | 5 (35.7) | 8 (27.6) | |
Values are expressed as mean±standard deviation, number (%), or median (interquartile range).
PPGL, pheochromocytoma and paraganglioma; NGS, next-generation sequencing; PCC, pheochromocytoma; PGL, paraganglioma.
a P value for the correlation between positive group and negative group;
b P value was calculated by t test, as they were parametric variables (Shapiro-Wilk test P=0.680 for age; P=0.574 for age of onset);
c Others include retroperitoneum, pancreas, para-aortic space, and bladder;
d P value was calculated by the Mann-Whitney U test, as it was a nonparametric variable (Shapiro-Wilk test P<0.001).
| Characteristic | Molecular clusters (total n=28) | P value | ||
|---|---|---|---|---|
|
|
||||
|
Pseudohypoxic TCA cycle-related (n=11) |
Pseudohypoxic VHL/EPAS1-related (n=6) |
Kinase-signaling (n=11) |
||
| Age, yra | 47.6±16.7 | 45.7±11.9 | 45.0±15.9 | 0.920 |
|
|
||||
| Age of onset, yra | 43.2±18.7 | 35.8±15.3 | 37.8±13.8 | 0.616 |
|
|
||||
| Female sex | 5 (45.5) | 4 (66.7) | 7 (63.6) | 0.599 |
|
|
||||
| Type of PPGL | ||||
| PCC | 4 (36.4) | 5 (83.3) | 11 (100.0) | 0.003 |
| PGL | 7 (63.6) | 1 (16.7) | 0 | |
|
|
||||
| Family history | 0 | 2 (33.3) | 7 (63.6) | 0.006 |
|
|
||||
| Location | ||||
| PCC (n=20) | ||||
| Unilateral adrenal gland | 4 (100.0) | 3 (60.0) | 7 (63.6) | 0.339 |
| Right adrenal gland | 2 (50.0) | 1 (20.0) | 5 (45.5) | |
| Left adrenal gland | 2 (50.0) | 2 (40.0) | 2 (18.2) | |
| Bilateral adrenal glands | 0 | 2 (40.0) | 4 (36.4) | |
| PGL (n=8) | ||||
| Head and neck | 1 (14.3) | 0 | 0 | 0.686 |
| Othersb | 6 (85.7) | 2 (100.0) | 0 | |
|
|
||||
| Tumor size, cm (n=23)c | 4.1 (3.1–4.7) | 3.5 (2.0–4.2) | 3.4 (2.0–4.7) | 0.688 |
|
|
||||
| Metastasis | 7 (63.6) | 3 (50.0) | 0 | 0.006 |
| Synchronous | 2 (18.2) | 2 (66.7) | 0 | 0.260 |
| Metachronous | 5 (45.5) | 1 (33.3) | 0 | |
|
|
||||
| Recurrence | 4 (36.4) | 2 (33.3) | 1 (9.1) | 0.292 |
|
|
||||
| Presence of other tumors | 1 (9.1) | 3 (50.0) | 8 (72.7) | 0.010 |
|
|
||||
| Discovery route (n=24) | ||||
| Incidental | 2 (25.0) | 5 (83.3) | 4 (40.0) | 0.086 |
| Symptomatic | 6 (75.0) | 1 (16.7) | 4 (40.0) | |
| Gene mutation | 0 | 0 | 2 (20.0) | |
|
|
||||
| Biochemical profile | ||||
| Adrenergic/Noradrenergic | 0 | 0 | 4 (36.4) | 0.036 |
| Adrenergic | 0 | 0 | 1 (9.1) | |
| Noradrenergic | 9 (81.8) | 3 (50.0) | 2 (18.2) | |
| Silent | 2 (18.2) | 3 (50.0) | 4 (36.4) | |
Values are expressed as mean±standard deviation, number (%), or median (interquartile range).
PPGL, pheochromocytoma and paraganglioma; TCA, tricarboxylic acid; VHL, von Hippel-Lindau; EPAS1, endothelial PAS domain-containing protein 1; PCC, pheochromocytoma; PGL, paraganglioma.
a P valuewas calculated by one-way analysis of variance, as they were parametric variables (Shapiro-Wilk test P=0.699 for age; P=0.461 for age of onset);
b Others include retroperitoneum, pancreas, para-aortic space, and bladder;
c P value was calculated by the Kruskal-Wallis test, as it was a nonparametric variable (Shapiro-Wilk test P<0.001).
PPGL, pheochromocytoma and paraganglioma; ACMG, American College of Medical Genetics and Genomics; PCC, pheochromocytoma; SDH, succinate dehydrogenase; NGS, next-generation sequencing; Rt, right; LP, likely pathogenic; Lt, left; VHL, von Hippel-Lindau; P, pathogenic; RCC, renal cell carcinoma; NET, neuroendocrine tumor; MAX, Myc-associated protein X; NF1, neurofibromatosis1; RET, rearranged during transformation; MTC, medullary thyroid carcinoma; PGL, paraganglioma; SCC, squamous cell carcinoma.
- 1. Lam AK. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr Pathol 2017;28:213–27.ArticlePubMed
- 2. McNichol AM. Differential diagnosis of pheochromocytomas and paragangliomas. Endocr Pathol 2001;12:407–15.ArticlePubMed
- 3. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer 2007;14:935–56.ArticlePubMed
- 4. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc 1983;58:802–4.PubMed
- 5. Johnson MH, Cavallo JA, Figenshau RS. Malignant and metastatic pheochromocytoma: case report and review of the literature. Urol Case Rep 2014;2:139–41.ArticlePubMedPMC
- 6. Bravo EL, Gifford RW Jr. Current concepts: pheochromocytoma: diagnosis, localization and management. N Engl J Med 1984;311:1298–303.ArticlePubMed
- 7. Manger WM, Gifford RW Jr, Hoffman BB. Pheochromocytoma: a clinical and experimental overview. Curr Probl Cancer 1985;9:1–89.
- 8. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015;11:101–11.ArticlePubMed
- 9. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014;14:108–19.ArticlePubMed
- 10. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009;94:1541–7.ArticlePubMed
- 11. Metzker ML. Sequencing technologies: the next generation. Nat Rev Genet 2010;11:31–46.ArticlePubMed
- 12. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000;287:848–51.ArticlePubMed
- 13. Nakamura K, Yamaki M, Sarada M, Nakayama S, Vibat CR, Gennis RB, et al. Two hydrophobic subunits are essential for the heme b ligation and functional assembly of complex II (succinate-ubiquinone oxidoreductase) from Escherichia coli. J Biol Chem 1996;271:521–7.ArticlePubMed
- 14. Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol 2015;2015:138573.ArticlePubMedPMC
- 15. Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev 2017;38:489–515.ArticlePubMedPMC
- 16. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014;99:1915–42.ArticlePubMed
- 17. NGS in PPGL (NGSnPPGL) Study Group. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol 2017;13:233–47.ArticlePubMed
- 18. Cascon A, Pita G, Burnichon N, Landa I, Lopez-Jimenez E, Montero-Conde C, et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J Clin Endocrinol Metab 2009;94:1701–5.ArticlePubMed
- 19. Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Brocker-Vriends AH, et al. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer 2001;31:274–81.ArticlePubMed
- 20. Kim KH, Chung JS, Kim WT, Oh CK, Chae YB, Yu HS, et al. Clinical experiences of pheochromocytoma in Korea. Yonsei Med J 2011;52:45–50.ArticlePubMed
- 21. Kim JH, Seong MW, Lee KE, Choi HJ, Ku EJ, Bae JH, et al. Germline mutations and genotype-phenotype correlations in patients with apparently sporadic pheochromocytoma/paraganglioma in Korea. Clin Genet 2014;86:482–6.ArticlePubMed
- 22. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016;174:G1–10.ArticlePubMed
- 23. Jochmanova I, Lazurova I. Diagnostika a manazment metastatickeho feochromocytomu a paragangliomu. Diagnosis and management of metastatic pheochromocytoma and paraganglioma. Vnitr Lek 2017;63:580–8.ArticlePubMed
- 24. Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2007;14:569–85.ArticlePubMed
- 25. Amar L, Fassnacht M, Gimenez-Roqueplo AP, Januszewicz A, Prejbisz A, Timmers H, et al. Long-term postoperative follow-up in patients with apparently benign pheochromocytoma and paraganglioma. Horm Metab Res 2012;44:385–9.ArticlePubMed
- 26. Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg 2006;141:1199–205.ArticlePubMed
- 27. Pacak K, Wimalawansa SJ. Pheochromocytoma and paraganglioma. Endocr Pract 2015;21:406–12.ArticlePubMed
- 28. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4–23.PubMedPMC
- 29. DeLelis RL, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumours of endocrine organs (IARC/World Health Organization Classification of Tumours); Lyon: IARC Press; 2004.
- 30. Rijken JA, Niemeijer ND, Jonker MA, Eijkelenkamp K, Jansen JC, van Berkel A, et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin Genet 2018;93:60–6.ArticlePubMed
- 31. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 2011;18:R253–76.ArticlePubMed
- 32. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002;346:1459–66.ArticlePubMed
- 33. Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: current approaches and future directions. Oncologist 2008;13:779–93.ArticlePubMed
- 34. Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 2007;92:3822–8.ArticlePubMed
- 35. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012;48:1739–49.ArticlePubMed
- 36. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 2003;63:5615–21.PubMed
- 37. Chen X, Hu L, Liu C, Ni G, Zhang Y. Tumor characteristics and surgical outcome in incidentally discovered pheochromocytomas and paragangliomas. Endocr Connect 2018;7:1142–9.ArticlePubMedPMC
- 38. Mariani-Costantini R. Paraganglioma: a multidisciplinary approach; Brisbane: Codon Publications; 2019.
- 39. Hoffman-Andrews L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci 2018;4:648–57.ArticlePubMedPMC
- 40. Geli J, Kiss N, Karimi M, Lee JJ, Backdahl M, Ekstrom TJ, et al. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: association to malignant behavior. Clin Cancer Res 2008;14:2551–9.ArticlePubMed
- 41. Margetts CD, Astuti D, Gentle DC, Cooper WN, Cascon A, Catchpoole D, et al. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel-Lindau and sporadic phaeochromocytomas. Endocr Relat Cancer 2005;12:161–72.ArticlePubMed
References
Figure & Data
References
Citations
- Patient Sex and Origin Influence Distribution of Driver Genes and Clinical Presentation of Paraganglioma
Susan Richter, Nicole Bechmann
Journal of the Endocrine Society.2024;[Epub] CrossRef - Novel and recurrent genetic variants of VHL, SDHB, and RET genes in Chinese pheochromocytoma and paraganglioma patients
Chong Li, Jingyi Li, Chao Han, Ting Wang, Lixia Zhang, Zhifang Wang, Tingting Wang, Lijun Xu, Guangzhao Qi, Guijun Qin, Xialian Li, Lili Zheng
Frontiers in Genetics.2023;[Epub] CrossRef - Genetic Study in Pheochromocytoma: Is It Possible to Stratify the Risk of Hereditary Pheochromocytoma?
Marta Araujo-Castro, César Mínguez Ojeda, Iñigo García Sanz, Maria Calatayud, Felicia Hanzu, Mireia Mora, Almudena Vicente, Concepción Blanco Carrera, Paz de Miguel Novoa, María del Carmen López García, Cristina Lamas, Laura Manjón-Miguélez, María del Cas
Neuroendocrinology.2023; 113(6): 657. CrossRef - The Role of VHL in the Development of von Hippel-Lindau Disease and Erythrocytosis
Petra Hudler, Mojca Urbancic
Genes.2022; 13(2): 362. CrossRef - Bilateral Pheochromocytoma with Germline MAX Variant without Family History
Shinnosuke Hata, Mai Asano, Hiroyuki Tominaga, Masahide Hamaguchi, Fumiya Hongo, Takeshi Usui, Eiichi Konishi, Michiaki Fukui
Clinics and Practice.2022; 12(3): 299. CrossRef - Diagnosis for Pheochromocytoma and Paraganglioma: A Joint Position Statement of the Korean Pheochromocytoma and Paraganglioma Task Force
Eu Jeong Ku, Kyoung Jin Kim, Jung Hee Kim, Mi Kyung Kim, Chang Ho Ahn, Kyung Ae Lee, Seung Hun Lee, You-Bin Lee, Kyeong Hye Park, Yun Mi Choi, Namki Hong, A Ram Hong, Sang-Wook Kang, Byung Kwan Park, Moon-Woo Seong, Myungshin Kim, Kyeong Cheon Jung, Chan
Endocrinology and Metabolism.2021; 36(2): 322. CrossRef - Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma
Masato Yonamine, Koichiro Wasano, Yuichi Aita, Takehito Sugasawa, Katsutoshi Takahashi, Yasushi Kawakami, Hitoshi Shimano, Hiroyuki Nishiyama, Hisato Hara, Mitsuhide Naruse, Takahiro Okamoto, Tadashi Matsuda, Shinji Kosugi, Kazuhiko Horiguchi, Akiyo Tanab
Cancers.2021; 13(16): 4014. CrossRef - Recurrent Germline Mutations of CHEK2 as a New Susceptibility Gene in Patients with Pheochromocytomas and Paragangliomas
Yinjie Gao, Chao Ling, Xiaosen Ma, Huiping Wang, Yunying Cui, Min Nie, Anli Tong, Dario De Biase
International Journal of Endocrinology.2021; 2021: 1. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite