
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 34(3); 2019 > Article
-
Review ArticleRecent Progress on Branched-Chain Amino Acids in Obesity, Diabetes, and Beyond
-
Md Abu Bakkar Siddik
 , Andrew C. Shin
, Andrew C. Shin -
Endocrinology and Metabolism 2019;34(3):234-246.
DOI: https://doi.org/10.3803/EnM.2019.34.3.234
Published online: September 26, 2019
Department of Nutritional Sciences, College of Human Sciences, Texas Tech University, Lubbock, TX, USA.
- Corresponding author: Andrew C. Shin. Department of Nutritional Sciences, College of Human Sciences, Texas Tech University, 2500 Broadway, Lubbock, TX 79409, USA. Tel: +1-806-834-1713, Fax: +1-806-742-2926, andrew.shin@ttu.edu
• Received: August 15, 2019 • Revised: September 16, 2019 • Accepted: September 21, 2019
Copyright © 2019 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Branched-chain amino acids (BCAAs) are essential amino acids that are not synthesized in our body; thus, they need to be obtained from food. They have shown to provide many physiological and metabolic benefits such as stimulation of pancreatic insulin secretion, milk production, adipogenesis, and enhanced immune function, among others, mainly mediated by mammalian target of rapamycin (mTOR) signaling pathway. After identified as a reliable marker of obesity and type 2 diabetes in recent years, an increasing number of studies have surfaced implicating BCAAs in the pathophysiology of other diseases such as cancers, cardiovascular diseases, and even neurodegenerative disorders like Alzheimer's disease. Here we discuss the most recent progress and review studies highlighting both correlational and potentially causative role of BCAAs in the development of these disorders. Although we are just beginning to understand the intricate relationships between BCAAs and some of the most prevalent chronic diseases, current findings raise a possibility that they are linked by a similar putative mechanism.
- Although first discovered and studied extensively since the late 1800s, it was the last 20 years or so during which branched-chain amino acids (BCAAs) have emerged as vital elements in various aspects of health and diseases as investigators began to unfold their roles beyond protein synthesis and degradation. BCAAs comprise leucine, isoleucine, and valine that have branched functional R groups [1]. Known as the essential amino acids due to the inability of animals to synthesize them (unlike microbial organisms and plants), BCAAs must be obtained through diet sources. All three BCAAs together make up about 20% of the total protein content and account for one-third of the dietary essential amino acids [2]. They are primarily known for their ability to promote protein synthesis and suppress proteolysis in both rodents and humans [3456789]. In support of these studies, BCAAs, specifically leucine, induce activation of mammalian target of rapamycin (mTOR) signaling pathway which is essential for initiation of protein synthesis [10111213]. This likely led to increased consumption of BCAAs as a performance-enhancing anabolic supplements by bodybuilders and individuals who would try to improve their physical fitness, although the effects of BCAAs per se that are independent of other essential amino acids or hormones are not clear [14]. BCAAs are also critical for protein/muscle preservation as evidenced by the beneficial effects of BCAA supplementation on protein turnover and muscle wasting in patients with cirrhosis [151617], kidney failure [18], liver cancers [19202122], and sepsis [232425]. Apart from their anabolic response, it has become clear that BCAAs serve as important nutrient signals and metabolic regulators. They can increase insulin secretion through their action at both protein and transcriptional levels in pancreatic β-cells [26], regulate adipose tissue metabolism [272829] and glucose homeostasis [3031], enhance intestinal development and health [3233], promote milk production from mammary gland [343536], participate in immune function [3738], and alter gut microbial diversity and functions [3940]. The metabolic and health benefits of BCAA ingestion have been described in detail in other excellent reviews [26414243].
- Based on ex vivo assays using extracts from various tissues, early studies have shown that unlike other amino acids, BCAAs first pass the liver due to the lack of BCAA aminotransferase (branched chain amino acid transaminase [BCAT]), the first enzyme in BCAA catabolic pathway [4445]. Instead, they are transaminated to branched-chain keto acids (BCKAs) by BCAT in organs such as muscle, kidney, heart, and adipose tissue, after which BCKAs are released back into circulation and taken up primarily by liver for oxidation through the rate-limiting enzyme complex, branched-chain α-keto acid dehydrogenase (BCKDH). The resulting acyl coenzyme A (acyl-CoA) are then further oxidized by different enzymes yielding either succinyl-CoA or acyl-CoA that can enter tricarboxylic acid cycle for adenosine triphosphate (ATP) synthesis. Interestingly, a recent study by Arany and Neinast [46] used an in vivo isotopic tracing to demonstrate that many peripheral organs do in fact oxidize BCAAs, indicating that tissues other than liver are fully capable of engaging in BCAA breakdown. While ingestion of excess glucose and fatty acids can be stored mainly in a form of glycogen in liver and muscle and triglycerides in white adipose tissue, amino acids including BCAAs are not converted to protein for later use, indicating that the only way to control BCAA overload is through its catabolic pathway. The clinical importance of BCAA catabolism is demonstrated in patients with inborn errors of metabolism such as maple syrup urine disease, a rare autosomal recessive disorder caused by mutations in BCKDH enzyme complex. The body is not able to break down BCAAs leading to supraphysiological levels of BCAAs and BCKAs in the blood. As a result, these individuals experience hypotonia and ketoacidosis as well as serious neurological issues such as developmental delay, hallucinations, seizures, and coma [47].
- In spite of BCAAs being the prerequisite for many health benefits as mentioned above, emerging studies in both animals and humans suggest an important role of BCAAs in the pathogenesis of metabolic disorders like obesity and diabetes, other chronic diseases such as cancer and heart disease, and even neurodegenerative disorders such as Alzheimer's disease (AD). The purpose of this review is to discuss and summarize recent advances in knowledge on both correlational and causal link between BCAAs and some of the most prevalent chronic diseases.
INTRODUCTION
- The observation of higher circulating BCAA levels in individuals with obesity was first reported by Felig et al. [48] nearly a half century ago, but the association between BCAAs and obesity was resurfaced only recently by She et al. [49] that was subsequently confirmed with high-throughput metabolomics analysis by other investigators [505152]. Once BCAAs gained traction, it did not take long for others to show that plasma BCAAs and/or their derived intermediates, BCKAs, are not just elevated in individuals with insulin resistance or type 2 diabetes [52535455], but they are also a strong predictive marker for future risk of these conditions regardless of age and ethnicity [5657585960]. Interestingly, mounting evidence suggests that BCAAs or BCKAs lead to hyperactivation of mTOR signaling [5061], induction of oxidative stress [62636465], mitochondrial dysfunction [6667], apoptosis [6869], and more importantly, insulin resistance and/or impaired glucose metabolism [507071727374757677], all of which are the key factors involved in the pathogenesis of diabetes. Consistent with these findings, BCAA supplementation with high-fat diet (HFD) or defective BCAA oxidation through deletion of methylmalonyl-CoA mutase in mice induce insulin resistance and impaired glucose tolerance [507577]. Jang et al. [74] have recently shown that 3-hydroxyisobutyrate, a catabolic intermediate of valine that is elevated in diabetic individuals, drives vascular fatty acid transport in muscle and causes glucose intolerance in mice. Findings from an in vitro study have demonstrated that a leucine metabolite, ketoisocaproic acid, suppresses insulin-stimulated glucose transport in L6 myotubes [76]. Conversely, depriving any one of BCAAs in regular chow diet or HFD in mice, or giving isocaloric and isonitrogenous diet with all three BCAAs restricted in genetically diabetic Zucker fatty rats improves insulin sensitivity and glycemic control [317879]. More recently, Cummings et al. [80] recapitulated these findings by showing that 80% restriction of BCAAs in western diet for 4 weeks significantly alleviates glucose intolerance and insulin resistance in mice. Mechanistically, drinking BCAA metabolite-containing water has been shown to decrease the active state of AKT (pAKT), a marker of insulin signaling, in muscle [74]. Newgard et al. [5081] have reported similar results in which BCAAs supplemented to HFD reduce pAKT and cause hyperactivation of mTOR signaling in skeletal muscle compared to BCAAs plus regular chow or HFD alone, leading to impaired glucose tolerance. In line with these findings, BCAA deprivation in mice increases pAKT in liver and improves insulin sensitivity [3178]. Together, these results support the notion of not just a correlative, but a causal role of BCAAs in the development of obesity, insulin resistance, and diabetes, possibly mediated by downregulation of AKT signaling pathway in insulin-sensitive tissues.
- An obvious question at this point is, why are circulating BCAA levels elevated in obese or insulin-resistant/diabetic individuals to begin with? The most intuitive answer would be that these individuals consume more food in general, thus naturally increasing their BCAA intake. However, it should be noted that higher BCAAs are observed even after matching protein/BCAA intake or overnight fasting [505457]. While it may be tempting to speculate that obese or diabetic individuals have higher BCAAs due to an increased proteolysis and/or decreased protein synthesis that is analogous to their elevated free fatty acids through increased lipid breakdown [8283], their whole-body protein synthesis and degradation do not seem to be different from those in healthy individuals [848586878889]. The most likely explanation is impaired BCAA catabolic pathway that would lead to BCAA build-up in the circulation. Indeed, mRNA of the rate-limiting enzyme BCKDH was significantly downregulated in subcutaneous adipose tissue of monozygotic twins discordant for obesity [90] and obese Pima Indians [29]. These findings are in agreement with our earlier study revealing reduced BCKDH activity in liver of obese macaques and humans [91], as well as others' findings with lower BCKDH protein expression in liver or subcutaneous adipose tissue of genetically obese ob/ob mice, diabetic fa/fa rats, or diet-induced obese mice [2949]. More recently, Zhou et al. [92] have beautifully demonstrated that treating either ob/ob or diet-induced obese mice with 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid (BT2), a pharmacological inhibitor of BCKDH kinase that lowers plasma BCAAs, significantly attenuated insulin resistance, indicating that efficient BCAA breakdown is critical for decreasing plasma BCAAs and improving insulin sensitivity.
- While many investigators have mainly focused on probing the cellular underpinnings through which BCAAs or their metabolites impair insulin signaling and glucose homeostasis, not much attention has been dedicated to their regulatory mechanism that might shed light on the dysfunctional BCAA degradation in obesity and diabetes. Early studies have shown that systemic hyperinsulinemia induced by either intravenous insulin injection or hyperinsulinemic euglycemic clamp lowers circulating BCAA levels in normal healthy subjects but less so in obese or diabetic individuals, implicating insulin resistance as a major cause for the elevated BCAAs in obesity and diabetes [93949596]. However, it was not clear how insulin is able to reduce plasma BCAAs. We hypothesized that this could be mediated primarily through induction of BCAA catabolism in liver, an organ with high BCKDH expression and activity [44]. Using hyperinsulinemic euglycemic clamps in rats, we demonstrated for the first time that insulin dose-dependently enhances BCKDH activity in liver but not white adipose tissue or muscle, and lowers plasma BCAA levels [91]. We further showed that this is accomplished by insulin action in the brain, specifically the mediobasal hypothalamus (MBH), a brain region critical for many physiological functions including the control of body weight, appetite, and nutrient partitioning [9798]. These results suggest that insulin resistance in the brain, not the periphery, may be the first event that triggers defective BCAA breakdown and the resultant increase in circulating BCAAs. Identifying the exact neuronal populations within the MBH responsible for the control of BCAA metabolism would be an important step toward better understanding of BCAA dysmetabolism in obesity and diabetes.
OBESITY AND TYPE 2 DIABETES
- Several independent labs have indicated a tight link between BCAAs and development of different types of cancers. Mayers et al. [99] have shown that plasma BCAAs are elevated in patients 2 to 5 years before they are diagnosed with pancreatic ductal adenocarcinoma (PDAC), increasing the risk by at least two-fold that is independent of intermediate development of diabetes. Similarly, mice bearing Kras-driven pancreatic tumors display higher plasma BCAAs before manifestation of subclinical cancer. This would presumably be associated with impaired glycemic control as commonly observed in patients with PDAC; however, there is no change in fasting glucose or glucose excursion during oral glucose tolerance test. These findings suggest that plasma BCAAs do not necessarily indicate impaired glycemic control or insulin resistance as in type 2 diabetes, but rather reflect an early consequence of PDAC. Using 13C-leucine to study BCAA uptake and incorporation into protein, the same group went on to demonstrate that BCAA metabolic fate is unique for specific cancers [100]. While PDAC tumors in mice have lower BCAA uptake, tumors derived from non-small cell lung carcinoma exhibit enhanced BCAA uptake and incorporation into protein. Other investigators have provided valuable insights into the role of BCAAs in proliferation and metabolic capacity of different types of cancers. Wang et al. [101] has shown that protein abundance of cytoplasmic BCAT1, the first enzyme in BCAA degradation pathway that is responsible for reversible transamination of BCAAs to BCKAs, is significantly upregulated in both low-malignant potential tumors and high-grade tumors from patients with epithelial ovarian cancer (EOC). Lentiviral short hairpin RNA (shRNA)-mediated knockdown of BCAT1 in EOC cell lines decreases the number of colony formation, and suppresses tumor cell replication and migration compared to tumor control cells. Interestingly, similar results are observed in tumors from hepatocellular carcinoma (HCC) [102] and breast cancer [103]. In addition to in vitro study, Zheng et al. [102] injected nude BALB/c mice subcutaneously with BCAT1-transfected HCC cells using lentiviral vector to demonstrate that BCAT1 overexpression in vivo accelerates tumor growth in mice. Support and extension of these findings comes from a recent study by Ericksen et al. [104] showing higher mRNAs of BCAT1/2 but lower BCKDH and downstream catabolic enzymes such as acyl-CoA dehydrogenase in tumors from HCC patients. Targeted metabolomics analysis in tumor versus adjacent non-tumor tissue revealed a significant increase in all three BCAAs but lower oxidized intermediates of BCAAs, indicating impaired BCAA breakdown in liver. Instead of being utilized as a source for ATP synthesis, it seems that BCAAs hyperactivate mTOR signaling in HCC tumors in both human and animals. Considering the role of mTOR in protein synthesis and cell proliferation [105], this explains the efficient growth rate of the tumor cells which is reversed after decreasing BCAAs in media or restoring BCAA breakdown. Conversely, treatment of HCC cells with lower BCAAs or mTOR inhibitor rapamycin, or either genetic or pharmacological inhibition of BCKDH kinase (BCKDH-suppressing enzyme) all results in significant reduction in tumor cell proliferation rates. It is interesting to note that tumors from breast cancer display gene upregulation of not only BCAT1 but also BCKDH and other downstream enzymes [103], indicating enhanced BCAA catabolism as opposed to impaired breakdown that is observed in HCC tumors described above (although whether or not the actual catabolic activity is increased is not clear). In spite of this difference in metabolic phenotype, similar to what occurs in EOC and HCC cells, BCAT1 knockdown decreases mTOR signaling and mitochondrial biogenesis, thereby repressing the growth rate of breast cancer cell lines [103]. Collectively, these findings clearly demonstrate a link between BCAA metabolism and tumor development across different types of cancer. While differences in BCAA metabolic fate are present between different cancers, BCAT1 seems to be a major common driver for tumor proliferation; thus, it may potentially serve as a therapeutic target for cancer treatment.
CANCERS
- Glucose and fatty acids as the primary energy substrates and their respective metabolism in the heart have been widely studied in cardiovascular health and disease states [106107]. On the other hand, the role of amino acids, specifically BCAAs, and its catabolic pathway in the control of cardiac function has been highlighted only recently. Ruiz-Canela et al. [108] have shown that baseline plasma levels of leucine and isoleucine, but not valine, are associated with increased risk of cardiovascular disease (CVD) at 1 year follow-up from the participants in the Prevención con Dieta Mediterránea (PREDIMED) study. Likewise, increased dietary BCAA intake in healthy individuals has shown to raise the hazard ratio for hypertension 3 years later, with 61% higher risk of incident hypertension after adjusting for variables such as age, sex, body mass index (BMI), and diabetes [109]. Importantly, in both studies, plasma BCAAs were measured after overnight fasting and dietary BCAA intake between the control and treatment groups were matched, hence eliminating a potentially significant confounder. Adding more evidence on circulating BCAAs as a biomarker, Du et al. [110] have revealed that plasma BCAAs are independent predictors for adverse cardiovascular events in patients with myocardial infarction and acute heart failure after adjusting for BMI and diabetes. While there seems to be a general agreement on the relationship between BCAAs and heart diseases, their potential causal effect was not examined until recently. Sun et al. [111] have shown that both BCAA and BCKA levels are higher in human failing hearts as well as pressure overload-induced mouse failing hearts that are associated with coordinated downregulation of key genes involved in BCAA catabolism including BCKDH. In light of their findings that suppression of BCKDH activity reduces cardiac systolic function in mice whereas BT2 treatment enhances cardiac BCAA degradation and preserves heart contractility, BCAA catabolic efficiency seems to play a vital role in preserving cardiac health after heart failure. Two other independent studies using either coronary artery ligation [112] or ischemia-reperfusion injury [113] were able to replicate these results, all pointing cardiac BCAA degradation as the prerequisite for proper heart remodeling after insults. While the exact mechanisms by which BCAAs or their defective breakdown exacerbate cardiac dysfunction are not clear, activated mTOR signaling and mitochondrial stress may be involved [111112]. It is also worth mentioning that in Dahl salt-sensitive rats that are fed a high-salt diet for induction of heart failure, BCAA supplementation in drinking water decreased heart rate and preserved cardiac function [114]. The discrepancy in BCAA effects may be due to the fact that this specific model leads to a progressive deterioration of heart function and a significant weight and muscle loss compared to the models described above.
HEART DISEASES
- BCAAs readily cross the blood-brain-barrier and compete for the same large amino acid transporter with aromatic amino acids (i.e., tyrosine, tryptophan, and phenylalanine) that serve as precursors for neurotransmitters involved in cognition and memory such as dopamine, serotonin, and norepinephrine [115116117]. Further, in the presence of α-ketoglutarate, the enzyme BCAT converts BCAAs to glutamate, an excitatory neurotransmitter that can induce excitotoxicity and neuronal death in AD brains [118]. This indicates that excess BCAAs may lead to a profound imbalance of these key neurotransmitters that are vital for proper neuronal functions and the resultant behaviors. Moreover, BCAA overload has been shown to induce neural oxidative stress and apoptosis [626364656869], and mTOR hyperactivation commonly induced by BCAAs can lead to insulin resistance in the brain [119120]. Importantly, all of these abnormalities represent the pathophysiological hallmarks of AD, making BCAAs a potentially significant contributor in the development of AD.
- Emerging evidence shows a clear link between BCAAs and AD. Hudd et al. [121] have demonstrated that serum BCAAs are elevated in subjects with mild cognitive impairment or AD compared to healthy controls. Interestingly, serum glutamate and the transaminating enzyme BCAT are also higher in AD patients and are positively correlated with AD severity. These results imply that glutamate overproduction through heightened BCAT activity may deteriorate brain functions and lead to AD, supporting the “glutamate excitotoxicity” hypothesis for AD pathogenesis. On the contrary, Gonzalez-Dominguez et al. [122] have shown that circulating valine levels are lower in AD patients compared to those in healthy subjects. Similarly, a prospective study by Tynkkynen et al. [123] have reported a significant association between lower serum valine levels and increased risk of AD, although this disappears after adjusting for BMI and cholesterol-lowering medications. Nonetheless, it is not clear why serum levels of other BCAAs (i.e., leucine and isoleucine) are not altered in these AD patients. Consistent with the concept of BCAAs being a contributor to AD development, through Mendelian randomization analysis a recent study has identified several single nucleotide polymorphisms related to isoleucine degradation in AD patients, including the gene PPM1K, a phosphatase that activates BCKDH enzyme complex [124]. These findings suggest for the first time that individuals genetically predisposed to higher BCAA levels may have increased susceptibility to AD.
- Rodent studies also support the role of BCAAs in AD pathogenesis. We have previously shown [125] that plasma BCAAs are already elevated in amyloid precursor protein/presenilin 1 (APP/PS1) transgenic AD mouse model at 5 months of age when overt cognitive deficit is not yet present [126127]. Our recent observations demonstrate that this increase is persistent at 9 months of age at which memory function is clearly impaired (unpublished), suggesting that BCAAs and/or their metabolites may serve as a potential predictive or diagnostic marker for AD. Li et al. [128] have recently shown that plasma BCAAs are higher in both aged and 3xTg AD mice, and this is associated with reduced expression of BCAT in the brain. The study further extends and shows that BCAA supplementation in drinking water stimulates Tau phosphorylation and exacerbates cognitive impairment in AD mice. Hyperphosphorylation of Tau is recapitulated following BCAT knockdown in neurons of AD mouse brains in mTOR-dependent manner [128]. In keeping with these results, 50% BCAA restriction significantly lowers plasma BCAAs and improves memory function in both regular chow and HFD-fed 3xTg AD mice [129]. Similarly, protein restriction cycling in 3xTg AD mice also has shown to improve memory function [130]. While these findings are consistent with the notion that BCAA overload or defective breakdown has a causal role in AD pathogenesis, several caveats need to be considered. First, since BCAT induces a reversible conversion between BCAAs and their keto acids, reduction of BCAT protein expression does not necessarily indicate impaired BCAA degradation. Examining the rate-limiting enzyme BCKDH and its actual oxidative activity would help address this issue. Second, dietary BCAA manipulation can alter caloric intake and body weight [50131] that can potentially influence the degree of brain pathology and consequently the magnitude of cognitive deficits in AD mice. Third, since BCAA supplementation or restriction in these studies leads to unequal calorie amount, and especially different nitrogen content in their diet that is essential for protein/DNA synthesis and enzymatic activities, it is difficult to make a clear conclusion. In future studies, it would be important to match caloric content and molar nitrogen quantity in diet in order to eliminate or minimize these potential confounders.
- It is interesting to note that dihydrolipoamide dehydrogenase (DLDH), or E3 subunit of mitochondrial proteins including BCKDH complex and pyruvate dehydrogenase, is in fact present in both human and mouse serum [132]. Yan et al. [132] have shown that unlike the one found in mitochondria, DLDH in serum is very sensitive to oxygen-induced inactivation and lacks the usual diaphorase activity for redox reactions. Considering that dysfunction of DLDH stimulates oxidative stress in both in vivo and in vitro [133134], whether this circulating subunit has different biological functions and whether its levels are altered and play a role in the development of AD, or any of the disease conditions aforementioned, needs to be investigated.
ALZHEIMER'S DISEASE
- Unlike glucose and lipids, studies on amino acids, more specifically BCAA metabolism and its physiological functions beyond protein synthesis, have garnered a substantial interest from different fields mainly in the last two decades. BCAAs have undoubtedly beneficial effects in preserving protein content in individuals with age-related and muscle-wasting conditions, and a number of studies have demonstrated their importance in immune function, adipogenesis, glucose uptake, and gut microbial diversity. Interestingly, recent studies from our lab as well as from other investigators have revealed that circulating BCAAs and/or their metabolites are elevated in prevalent chronic diseases such as obesity, type 2 diabetes, cancers, CVDs, and neurodegenerative disorders like AD (Fig. 1). These similar findings across different disease conditions, while novel and exciting, are not all that surprising given that they are risk factors for one another and share several key physiological dysfunctions including insulin resistance, inflammation, and impaired nutrient partitioning. Although BCAA levels alone most likely will not be able to serve as a diagnostic marker specifically for any one of these diseases for the very findings discussed above, perhaps they might be utilized in conjunction with other existing biomarkers to enhance diagnostic precision or to facilitate early detection. Current evidence points to hyperactivated mTOR signaling as the critical mediator in some of the diseases. Clearly, more mechanistic and well-controlled studies are necessary to confirm current results and gain deeper understanding of how BCAAs become elevated in these chronic illnesses with common abnormal features, and provide insights into how they contribute to specific pathologies and symptoms. Once future studies reproduce and validate the causal role of BCAAs, it would be important to consider two potential therapeutic strategies to fight these disease conditions: (1) assess a safe limit of BCAA intake in individuals that already have or are susceptible to the diseases in order to determine appropriate dietary BCAA recommendations and (2) establish pharmacological interventions that can ameliorate BCAA-driven impairment in cellular signaling and subsequent maladaptive phenotypes.
CONCLUSIONS
-
Acknowledgements
- Research of Dr. Andrew C. Shin was funded by the NIH (DK099463) and Wylie Briscoe Fund from Texas Tech University. We would like to thank Jungwon Shin for technical help on the summary figure.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Chou PY, Fasman GD. Structural and functional role of leucine residues in proteins. J Mol Biol 1973;74:263–281. ArticlePubMed
- 2. Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr 1984;4:409–454. ArticlePubMed
- 3. Bukhari SS, Phillips BE, Wilkinson DJ, Limb MC, Rankin D, Mitchell WK, et al. Intake of low-dose leucine-rich essential amino acids stimulates muscle anabolism equivalently to bolus whey protein in older women at rest and after exercise. Am J Physiol Endocrinol Metab 2015;308:E1056–E1065. ArticlePubMed
- 4. Buse MG. In vivo effects of branched chain amino acids on muscle protein synthesis in fasted rats. Horm Metab Res 1981;13:502–505. ArticlePubMedPDF
- 5. Garlick PJ, Grant I. Amino acid infusion increases the sensitivity of muscle protein synthesis in vivo to insulin. Effect of branched-chain amino acids. Biochem J 1988;254:579–584. ArticlePubMedPMCPDF
- 6. Kobayashi H, Kato H, Hirabayashi Y, Murakami H, Suzuki H. Modulations of muscle protein metabolism by branched-chain amino acids in normal and muscle-atrophying rats. J Nutr 2006;136(1 Suppl):234S–236S. ArticlePubMedPDF
- 7. Li JB, Jefferson LS. Influence of amino acid availability on protein turnover in perfused skeletal muscle. Biochim Biophys Acta 1978;544:351–359. ArticlePubMed
- 8. Louard RJ, Barrett EJ, Gelfand RA. Overnight branched-chain amino acid infusion causes sustained suppression of muscle proteolysis. Metabolism 1995;44:424–429. ArticlePubMed
- 9. Matthews DE. Observations of branched-chain amino acid administration in humans. J Nutr 2005;135(6 Suppl):1580S–1584S. ArticlePubMedPMCPDF
- 10. Buse MG, Reid SS. Leucine. A possible regulator of protein turnover in muscle. J Clin Invest 1975;56:1250–1261. ArticlePubMedPMC
- 11. Hong SO, Layman DK. Effects of leucine on in vitro protein synthesis and degradation in rat skeletal muscles. J Nutr 1984;114:1204–1212. ArticlePubMedPDF
- 12. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274–293. ArticlePubMedPMC
- 13. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016;351:43–48. ArticlePubMed
- 14. Wolfe RR. Branched-chain amino acids and muscle protein synthesis in humans: myth or reality? J Int Soc Sports Nutr 2017;14:30. ArticlePubMedPMCPDF
- 15. Chin SE, Shepherd RW, Thomas BJ, Cleghorn GJ, Patrick MK, Wilcox JA, et al. Nutritional support in children with end-stage liver disease: a randomized crossover trial of a branched-chain amino acid supplement. Am J Clin Nutr 1992;56:158–163. ArticlePubMedPDF
- 16. Habu D, Nishiguchi S, Nakatani S, Kawamura E, Lee C, Enomoto M, et al. Effect of oral supplementation with branched-chain amino acid granules on serum albumin level in the early stage of cirrhosis: a randomized pilot trial. Hepatol Res 2003;25:312–318. ArticlePubMed
- 17. Tsien C, Davuluri G, Singh D, Allawy A, Ten Have GA, Thapaliya S, et al. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 2015;61:2018–2029. ArticlePubMedPMC
- 18. Tietze IN, Pedersen EB. Effect of fish protein supplementation on aminoacid profile and nutritional status in haemodialysis patients. Nephrol Dial Transplant 1991;6:948–954. ArticlePubMedPDF
- 19. Choudry HA, Pan M, Karinch AM, Souba WW. Branched-chain amino acid-enriched nutritional support in surgical and cancer patients. J Nutr 2006;136(1 Suppl):314S–318S. ArticlePubMedPDF
- 20. Ishihara T, Iwasa M, Tanaka H, Kaito M, Ikoma J, Shibata T, et al. Effect of branched-chain amino acids in patients receiving intervention for hepatocellular carcinoma. World J Gastroenterol 2014;20:2673–2680. ArticlePubMedPMC
- 21. Kakazu E, Kondo Y, Kogure T, Ninomiya M, Kimura O, Iwata T, et al. Supplementation of branched-chain amino acids maintains the serum albumin level in the course of hepatocellular carcinoma recurrence. Tohoku J Exp Med 2013;230:191–196. ArticlePubMed
- 22. Togo S, Tanaka K, Morioka D, Sugita M, Ueda M, Miura Y, et al. Usefulness of granular BCAA after hepatectomy for liver cancer complicated with liver cirrhosis. Nutrition 2005;21:480–486. ArticlePubMed
- 23. Bower RH, Muggia-Sullam M, Vallgren S, Hurst JM, Kern KA, LaFrance R, et al. Branched chain amino acid-enriched solutions in the septic patient. A randomized, prospective trial. Ann Surg 1986;203:13–20. ArticlePubMed
- 24. Garcia-de-Lorenzo A, Ortiz-Leyba C, Planas M, Montejo JC, Nunez R, Ordonez FJ, et al. Parenteral administration of different amounts of branch-chain amino acids in septic patients: clinical and metabolic aspects. Crit Care Med 1997;25:418–424. ArticlePubMed
- 25. Jimenez Jimenez FJ, Ortiz Leyba C, Morales Menedez S, Barros Perez M, Munoz Garcia J. Prospective study on the efficacy of branched-chain amino acids in septic patients. JPEN J Parenter Enteral Nutr 1991;15:252–261. ArticlePubMed
- 26. Yang J, Chi Y, Burkhardt BR, Guan Y, Wolf BA. Leucine metabolism in regulation of insulin secretion from pancreatic beta cells. Nutr Rev 2010;68:270–279. ArticlePubMedPMCPDF
- 27. Estrada-Alcalde I, Tenorio-Guzman MR, Tovar AR, Salinas-Rubio D, Torre-Villalvazo I, Torres N, et al. Metabolic fate of branched-chain amino acids during adipogenesis, in adipocytes from obese mice and C2C12 myotubes. J Cell Biochem 2017;118:808–818. ArticlePubMed
- 28. Wallace M, Green CR, Roberts LS, Lee YM, McCarville JL, Sanchez-Gurmaches J, et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat Chem Biol 2018;14:1021–1031. ArticlePubMedPMCPDF
- 29. Lackey DE, Lynch CJ, Olson KC, Mostaedi R, Ali M, Smith WH, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab 2013;304:E1175–E1187. ArticlePubMedPMC
- 30. Doi M, Yamaoka I, Fukunaga T, Nakayama M. Isoleucine, a potent plasma glucose-lowering amino acid, stimulates glucose uptake in C2C12 myotubes. Biochem Biophys Res Commun 2003;312:1111–1117. ArticlePubMed
- 31. Xiao F, Yu J, Guo Y, Deng J, Li K, Du Y, et al. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014;63:841–850. ArticlePubMed
- 32. Chang Y, Cai H, Liu G, Chang W, Zheng A, Zhang S, et al. Effects of dietary leucine supplementation on the gene expression of mammalian target of rapamycin signaling pathway and intestinal development of broilers. Anim Nutr 2015;1:313–319. ArticlePubMedPMC
- 33. Zhang S, Qiao S, Ren M, Zeng X, Ma X, Wu Z, et al. Supplementation with branched-chain amino acids to a low-protein diet regulates intestinal expression of amino acid and peptide transporters in weanling pigs. Amino Acids 2013;45:1191–1205. ArticlePubMedPDF
- 34. Doelman J, Kim JJ, Carson M, Metcalf JA, Cant JP. Branched-chain amino acid and lysine deficiencies exert different effects on mammary translational regulation. J Dairy Sci 2015;98:7846–7855. ArticlePubMed
- 35. Lei J, Feng D, Zhang Y, Zhao FQ, Wu Z, San Gabriel A, et al. Nutritional and regulatory role of branched-chain amino acids in lactation. Front Biosci (Landmark Ed) 2012;17:2725–2739. ArticlePubMed
- 36. Strathe AV, Bruun TS, Zerrahn JE, Tauson AH, Hansen CF. The effect of increasing the dietary valine-to-lysine ratio on sow metabolism, milk production, and litter growth. J Anim Sci 2016;94:155–164. ArticlePubMedPDF
- 37. Mao X, Qi S, Yu B, He J, Yu J, Chen D. Zn(2+) and L-isoleucine induce the expressions of porcine β-defensins in IPEC-J2 cells. Mol Biol Rep 2013;40:1547–1552. ArticlePubMedPDF
- 38. Rivas-Santiago CE, Rivas-Santiago B, Leon DA, Castaneda-Delgado J, Hernandez Pando R. Induction of β-defensins by l-isoleucine as novel immunotherapy in experimental murine tuberculosis. Clin Exp Immunol 2011;164:80–89. ArticlePubMedPMC
- 39. Neis EP, Dejong CH, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015;7:2930–2946. ArticlePubMedPMC
- 40. Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016;535:376–381. ArticlePubMedPDF
- 41. Zhang S, Zeng X, Ren M, Mao X, Qiao S. Novel metabolic and physiological functions of branched chain amino acids: a review. J Anim Sci Biotechnol 2017;8:10. ArticlePubMedPMCPDF
- 42. Holecek M. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab (Lond) 2018;15:33. ArticlePubMedPMCPDF
- 43. Bifari F, Nisoli E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: a pharmacological point of view. Br J Pharmacol 2017;174:1366–1377. ArticlePubMed
- 44. Hutson SM, Sweatt AJ, Lanoue KF. Branched-chain [corrected] amino acid metabolism: implications for establishing safe intakes. J Nutr 2005;135(6 Suppl):1557S–1564S. ArticlePubMedPDF
- 45. Shimomura Y, Obayashi M, Murakami T, Harris RA. Regulation of branched-chain amino acid catabolism: nutritional and hormonal regulation of activity and expression of the branched-chain alpha-keto acid dehydrogenase kinase. Curr Opin Clin Nutr Metab Care 2001;4:419–423. ArticlePubMed
- 46. Arany Z, Neinast M. Branched chain amino acids in metabolic disease. Curr Diab Rep 2018;18:76. ArticlePubMedPDF
- 47. Blackburn PR, Gass JM, Vairo FPE, Farnham KM, Atwal HK, Macklin S, et al. Maple syrup urine disease: mechanisms and management. Appl Clin Genet 2017;10:57–66. ArticlePubMedPMC
- 48. Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 1969;281:811–816. ArticlePubMed
- 49. She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab 2007;293:E1552–E1563. ArticlePubMedPMC
- 50. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9:311–326. ArticlePubMedPMC
- 51. Kim JY, Park JY, Kim OY, Ham BM, Kim HJ, Kwon DY, et al. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS). J Proteome Res 2010;9:4368–4375. ArticlePubMed
- 52. Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010;18:1695–1700. ArticlePubMedPMC
- 53. Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 2009;139:1073–1081. ArticlePubMedPMCPDF
- 54. Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ, Goh DL, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 2010;53:757–767. ArticlePubMedPMCPDF
- 55. Shi L, Brunius C, Lehtonen M, Auriola S, Bergdahl IA, Rolandsson O, et al. Plasma metabolites associated with type 2 diabetes in a Swedish population: a case-control study nested in a prospective cohort. Diabetologia 2018;61:849–861. ArticlePubMedPMCPDF
- 56. Trico D, Prinsen H, Giannini C, de Graaf R, Juchem C, Li F, et al. Elevated α-hydroxybutyrate and branched-chain amino acid levels predict deterioration of glycemic control in adolescents. J Clin Endocrinol Metab 2017;102:2473–2481. ArticlePubMedPMCPDF
- 57. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448–453. ArticlePubMedPMCPDF
- 58. Lu Y, Wang Y, Ong CN, Subramaniam T, Choi HW, Yuan JM, et al. Metabolic signatures and risk of type 2 diabetes in a Chinese population: an untargeted metabolomics study using both LC-MS and GC-MS. Diabetologia 2016;59:2349–2359. ArticlePubMedPDF
- 59. Wurtz P, Soininen P, Kangas AJ, Ronnemaa T, Lehtimaki T, Kahonen M, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 2013;36:648–655. ArticlePubMedPMC
- 60. McCormack SE, Shaham O, McCarthy MA, Deik AA, Wang TJ, Gerszten RE, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes 2013;8:52–61. ArticlePubMed
- 61. Jeganathan S, Abdullahi A, Zargar S, Maeda N, Riddell MC, Adegoke OA. Amino acid-induced impairment of insulin sensitivity in healthy and obese rats is reversible. Physiol Rep 2014;2:e12067. ArticlePubMedPMC
- 62. Funchal C, Latini A, Jacques-Silva MC, Dos Santos AQ, Buzin L, Gottfried C, et al. Morphological alterations and induction of oxidative stress in glial cells caused by the branched-chain alpha-keto acids accumulating in maple syrup urine disease. Neurochem Int 2006;49:640–650. ArticlePubMed
- 63. Bridi R, Braun CA, Zorzi GK, Wannmacher CM, Wajner M, Lissi EG, et al. Alpha-keto acids accumulating in maple syrup urine disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab Brain Dis 2005;20:155–167. ArticlePubMedPDF
- 64. Lu G, Sun H, She P, Youn JY, Warburton S, Ping P, et al. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest 2009;119:1678–1687. ArticlePubMedPMC
- 65. Oyarzabal A, Martinez-Pardo M, Merinero B, Navarrete R, Desviat LR, Ugarte M, et al. A novel regulatory defect in the branched-chain α-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. Hum Mutat 2013;34:355–362. ArticlePubMed
- 66. Amaral AU, Leipnitz G, Fernandes CG, Seminotti B, Schuck PF, Wajner M. Alpha-ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res 2010;1324:75–84. ArticlePubMed
- 67. Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, et al. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev 2007;21:784–796. ArticlePubMedPMC
- 68. Jouvet P, Rustin P, Taylor DL, Pocock JM, Felderhoff-Mueser U, Mazarakis ND, et al. Branched chain amino acids induce apoptosis in neural cells without mitochondrial membrane depolarization or cytochrome c release: implications for neurological impairment associated with maple syrup urine disease. Mol Biol Cell 2000;11:1919–1932. ArticlePubMedPMC
- 69. Jouvet P, Kozma M, Mehmet H. Primary human fibroblasts from a maple syrup urine disease patient undergo apoptosis following exposure to physiological concentrations of branched chain amino acids. Ann N Y Acad Sci 2000;926:116–121. ArticlePubMed
- 70. Balage M, Dupont J, Mothe-Satney I, Tesseraud S, Mosoni L, Dardevet D. Leucine supplementation in rats induced a delay in muscle IR/PI3K signaling pathway associated with overall impaired glucose tolerance. J Nutr Biochem 2011;22:219–226. ArticlePubMed
- 71. Nicastro H, Zanchi NE, da Luz CR, de Moraes WM, Ramona P, de Siqueira Filho MA, et al. Effects of leucine supplementation and resistance exercise on dexamethasone-induced muscle atrophy and insulin resistance in rats. Nutrition 2012;28:465–471. ArticlePubMed
- 72. Zanchi NE, Guimaraes-Ferreira L, de Siqueira-Filho MA, Felitti V, Nicastro H, Bueno C, et al. Dose and latency effects of leucine supplementation in modulating glucose homeostasis: opposite effects in healthy and glucocorticoid-induced insulin-resistance states. Nutrients 2012;4:1851–1867. ArticlePubMedPMC
- 73. Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes 2010;59:2426–2434. ArticlePubMedPMC
- 74. Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 2016;22:421–426. ArticlePubMedPMCPDF
- 75. Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab 2016;5:926–936. ArticlePubMedPMC
- 76. Moghei M, Tavajohi-Fini P, Beatty B, Adegoke OA. Ketoisocaproic acid, a metabolite of leucine, suppresses insulin-stimulated glucose transport in skeletal muscle cells in a BCAT2-dependent manner. Am J Physiol Cell Physiol 2016;311:C518–C527. ArticlePubMedPMC
- 77. Zhang F, Zhao S, Yan W, Xia Y, Chen X, Wang W, et al. Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine 2016;13:157–167. ArticlePubMedPMC
- 78. Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 2011;60:746–756. ArticlePubMedPMC
- 79. White PJ, Lapworth AL, An J, Wang L, McGarrah RW, Stevens RD, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab 2016;5:538–551. ArticlePubMedPMC
- 80. Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol 2018;596:623–645. ArticlePubMed
- 81. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 2012;15:606–614. ArticlePubMedPMC
- 82. Fabbrini E, Magkos F, Conte C, Mittendorfer B, Patterson BW, Okunade AL, et al. Validation of a novel index to assess insulin resistance of adipose tissue lipolytic activity in obese subjects. J Lipid Res 2012;53:321–324. ArticlePubMedPMC
- 83. Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, et al. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 1989;84:205–213. ArticlePubMedPMC
- 84. Barazzoni R, Kiwanuka E, Zanetti M, Cristini M, Vettore M, Tessari P. Insulin acutely increases fibrinogen production in individuals with type 2 diabetes but not in individuals without diabetes. Diabetes 2003;52:1851–1856. ArticlePubMed
- 85. Tessari P, Coracina A, Kiwanuka E, Vedovato M, Vettore M, Valerio A, et al. Effects of insulin on methionine and homocysteine kinetics in type 2 diabetes with nephropathy. Diabetes 2005;54:2968–2976. ArticlePubMed
- 86. Halvatsiotis PG, Turk D, Alzaid A, Dinneen S, Rizza RA, Nair KS. Insulin effect on leucine kinetics in type 2 diabetes mellitus. Diabetes Nutr Metab 2002;15:136–142. PubMed
- 87. Luzi L, Petrides AS, De Fronzo RA. Different sensitivity of glucose and amino acid metabolism to insulin in NIDDM. Diabetes 1993;42:1868–1877. ArticlePubMed
- 88. Biolo G, Tessari P, Inchiostro S, Bruttomesso D, Sabadin L, Fongher C, et al. Fasting and postmeal phenylalanine metabolism in mild type 2 diabetes. Am J Physiol 1992;263(5 Pt 1):E877–E883. ArticlePubMed
- 89. Tessari P, Cecchet D, Cosma A, Puricelli L, Millioni R, Vedovato M, et al. Insulin resistance of amino acid and protein metabolism in type 2 diabetes. Clin Nutr 2011;30:267–272. ArticlePubMed
- 90. Pietilainen KH, Naukkarinen J, Rissanen A, Saharinen J, Ellonen P, Keranen H, et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: pathways behind acquired obesity. PLoS Med 2008;5:e51. ArticlePubMedPMC
- 91. Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab 2014;20:898–909. ArticlePubMedPMC
- 92. Zhou M, Jing S, Wu CY, Shu L, Dong W, Liu Y, et al. Targeting BCAA catabolism to treat obesity-associated insulin resistance. Diabetes 2019;68:1730–1746. ArticlePubMedPMC
- 93. Schauder P, Schroder K, Matthaei D, Henning HV, Langenbeck U. Influence of insulin on blood levels of branched chain keto and amino acids in man. Metabolism 1983;32:323–327. ArticlePubMed
- 94. Tessari P, Nosadini R, Trevisan R, De Kreutzenberg SV, Inchiostro S, Duner E, et al. Defective suppression by insulin of leucine-carbon appearance and oxidation in type 1, insulin-dependent diabetes mellitus. Evidence for insulin resistance involving glucose and amino acid metabolism. J Clin Invest 1986;77:1797–1804. ArticlePubMedPMC
- 95. Caballero B, Wurtman RJ. Differential effects of insulin resistance on leucine and glucose kinetics in obesity. Metabolism 1991;40:51–58. ArticlePubMed
- 96. Forlani G, Vannini P, Marchesini G, Zoli M, Ciavarella A, Pisi E. Insulin-dependent metabolism of branched-chain amino acids in obesity. Metabolism 1984;33:147–150. ArticlePubMed
- 97. Sohn JW. Network of hypothalamic neurons that control appetite. BMB Rep 2015;48:229–233. ArticlePubMedPMCPDF
- 98. Rui L. Brain regulation of energy balance and body weight. Rev Endocr Metab Disord 2013;14:387–407. ArticlePubMedPDF
- 99. Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 2014;20:1193–1198. ArticlePubMedPMCPDF
- 100. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016;353:1161–1165. ArticlePubMedPMC
- 101. Wang ZQ, Faddaoui A, Bachvarova M, Plante M, Gregoire J, Renaud MC, et al. BCAT1 expression associates with ovarian cancer progression: possible implications in altered disease metabolism. Oncotarget 2015;6:31522–31543. ArticlePubMedPMC
- 102. Zheng YH, Hu WJ, Chen BC, Grahn TH, Zhao YR, Bao HL, et al. BCA.T1, a key prognostic predictor of hepatocellular carcinoma, promotes cell proliferation and induces chemoresistance to cisplatin. Liver Int 2016;36:1836–1847. ArticlePubMed
- 103. Zhang L, Han J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem Biophys Res Commun 2017;486:224–231. ArticlePubMed
- 104. Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, et al. Loss of BCAA catabolism during carcinogenesis enhances mTORC1 activity and promotes tumor development and progression. Cell Metab 2019;29:1151–1165. ArticlePubMedPMC
- 105. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004;18:1926–1945. ArticlePubMed
- 106. Hunter WG, Kelly JP, McGarrah RW 3rd, Kraus WE, Shah SH. Metabolic dysfunction in heart failure: diagnostic, prognostic, and pathophysiologic insights from metabolomic profiling. Curr Heart Fail Rep 2016;13:119–131. ArticlePubMedPMCPDF
- 107. Wang P, Xu L, Sun A. Energy remodeling, mitochondrial disorder and heart failure. Curr Pharm Des 2016;22:4823–4829. ArticlePubMed
- 108. Ruiz-Canela M, Toledo E, Clish CB, Hruby A, Liang L, Salas-Salvado J, et al. Plasma branched-chain amino acids and incident cardiovascular disease in the PREDIMED trial. Clin Chem 2016;62:582–592. ArticlePubMedPMCPDF
- 109. Mirmiran P, Teymoori F, Asghari G, Azizi F. Dietary intakes of branched chain amino acids and the incidence of hypertension: a population-based prospective cohort study. Arch Iran Med 2019;22:182–188. PubMed
- 110. Du X, Li Y, Wang Y, You H, Hui P, Zheng Y, et al. Increased branched-chain amino acid levels are associated with long-term adverse cardiovascular events in patients with STEMI and acute heart failure. Life Sci 2018;209:167–172. ArticlePubMed
- 111. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 2016;133:2038–2049. ArticlePubMedPMC
- 112. Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, et al. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 2016;311:H1160–H1169. ArticlePubMed
- 113. Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab 2017;25:374–385. ArticlePubMedPMC
- 114. Tanada Y, Shioi T, Kato T, Kawamoto A, Okuda J, Kimura T. Branched-chain amino acids ameliorate heart failure with cardiac cachexia in rats. Life Sci 2015;137:20–27. ArticlePubMed
- 115. Fernstrom JD. Large neutral amino acids: dietary effects on brain neurochemistry and function. Amino Acids 2013;45:419–430. ArticlePubMedPDF
- 116. Fernstrom JD. Branched-chain amino acids and brain function. J Nutr 2005;135(6 Suppl):1539S–1546S. ArticlePubMedPDF
- 117. Pardridge WM. Kinetics of competitive inhibition of neutral amino acid transport across the blood-brain barrier. J Neurochem 1977;28:103–108. ArticlePubMed
- 118. Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer's disease. J Alzheimers Dis 2017;57:1041–1048. ArticlePubMedPMC
- 119. Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest 2008;118:2959–2968. ArticlePubMedPMC
- 120. Lee HK, Kwon B, Lemere CA, de la Monte S, Itamura K, Ha AY, et al. mTORC2 (Rictor) in Alzheimer's disease and reversal of amyloid-β expression-induced insulin resistance and toxicity in rat primary cortical neurons. J Alzheimers Dis 2017;56:1015–1036. ArticlePubMedPMC
- 121. Hudd F, Shiel A, Harris M, Bowdler P, McCann B, Tsivos D, et al. Novel blood biomarkers that correlate with cognitive performance and hippocampal volumetry: potential for early diagnosis of Alzheimer's disease. J Alzheimers Dis 2019;67:931–947. ArticlePubMed
- 122. Gonzalez-Dominguez R, Garcia-Barrera T, Gomez-Ariza JL. Metabolite profiling for the identification of altered metabolic pathways in Alzheimer's disease. J Pharm Biomed Anal 2015;107:75–81. ArticlePubMed
- 123. Tynkkynen J, Chouraki V, van der Lee SJ, Hernesniemi J, Yang Q, Li S, et al. Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer's disease: a prospective study in eight cohorts. Alzheimers Dement 2018;14:723–733. ArticlePubMedPMC
- 124. Larsson SC, Markus HS. Branched-chain amino acids and Alzheimer's disease: a Mendelian randomization analysis. Sci Rep 2017;7:13604. ArticlePubMedPMC
- 125. Ruiz HH, Chi T, Shin AC, Lindtner C, Hsieh W, Ehrlich M, et al. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer's disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimers Dement 2016;12:851–861. ArticlePubMedPMC
- 126. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006;7:940–946. ArticlePubMedPMC
- 127. Knight EM, Ruiz HH, Kim SH, Harte JC, Hsieh W, Glabe C, et al. Unexpected partial correction of metabolic and behavioral phenotypes of Alzheimer's APP/PSEN1 mice by gene targeting of diabetes/Alzheimer's-related Sorcs1. Acta Neuropathol Commun 2016;4:16. ArticlePubMedPMC
- 128. Li H, Ye D, Xie W, Hua F, Yang Y, Wu J, et al. Defect of branched-chain amino acid metabolism promotes the development of Alzheimer's disease by targeting the mTOR signaling. Biosci Rep 2018;38:BSR20180127. ArticlePubMedPMC
- 129. Tournissac M, Vandal M, Tremblay C, Bourassa P, Vancassel S, Emond V, et al. Dietary intake of branched-chain amino acids in a mouse model of Alzheimer's disease: effects on survival, behavior, and neuropathology. Alzheimers Dement (N Y) 2018;4:677–687. ArticlePubMedPMC
- 130. Parrella E, Maxim T, Maialetti F, Zhang L, Wan J, Wei M, et al. Protein restriction cycles reduce IGF-1 and phosphorylated Tau, and improve behavioral performance in an Alzheimer's disease mouse model. Aging Cell 2013;12:257–268. ArticlePubMedPMC
- 131. Du Y, Meng Q, Zhang Q, Guo F. Isoleucine or valine deprivation stimulates fat loss via increasing energy expenditure and regulating lipid metabolism in WAT. Amino Acids 2012;43:725–734. ArticlePubMedPDF
- 132. Yan LJ, Thangthaeng N, Sumien N, Forster MJ. Serum dihydrolipoamide dehydrogenase is a labile enzyme. J Biochem Pharmacol Res 2013;1:30–42. PubMedPMC
- 133. Vaubel RA, Rustin P, Isaya G. Mutations in the dimer interface of dihydrolipoamide dehydrogenase promote site-specific oxidative damages in yeast and human cells. J Biol Chem 2011;286:40232–40245. ArticlePubMedPMC
- 134. Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L, et al. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem 2004;88:1352–1360. ArticlePubMed
References
Fig. 1
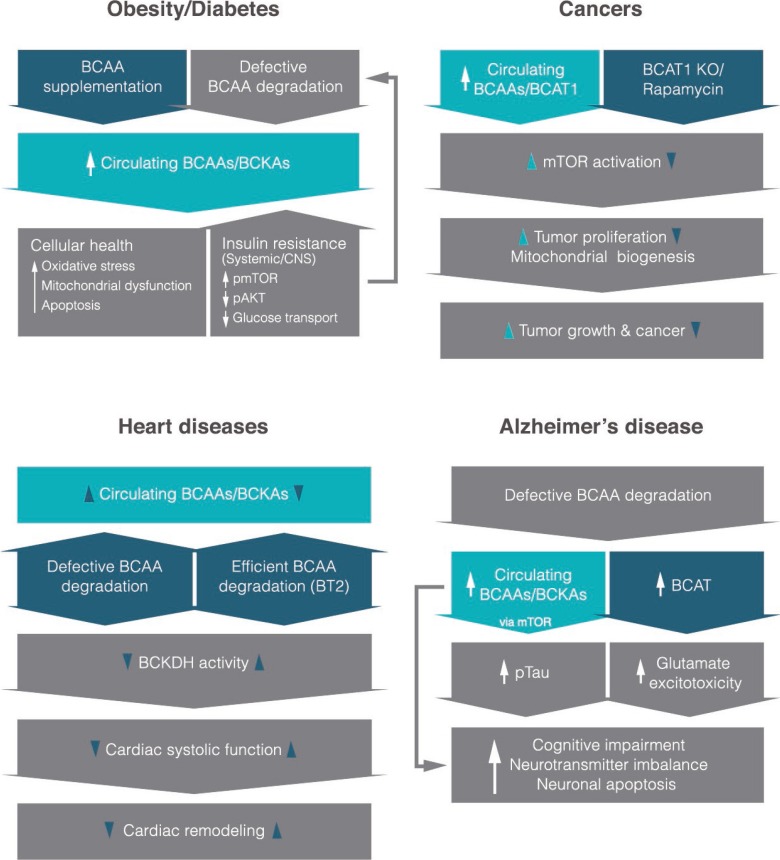
An overview of what is currently known about the role of branched-chain amino acids (BCAAs) or their derived keto-acids (i.e., branched-chain keto acids [BCKAs]) in different disease states and their potential molecular mechanisms. It is interesting to note that most studies reveal higher circulating levels of BCAAs in each disease. Items in dark blue shapes indicate mechanistic interventions based on recent studies. CNS, central nervous system; pmTOR, phosphorylated mammalian target of rapamycin; pAKT, phosphorylated protein kinase B; BCAT1, cytoplasmic branched-chain amino acid transaminase; KO, knockout; BT2, 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid; BCKDH, branched-chain α-keto acid dehydrogenase.
Figure & Data
References
Citations
Citations to this article as recorded by 
- First trimester metabolomics 1H-NMR study of the urinary profile predicts gestational diabetes mellitus development in obese women
Cristina Piras, Isabella Neri, Roberta Pintus, Antonio Noto, Elisabetta Petrella, Francesca Monari, Angelica Dessì, Vassilios Fanos, Luigi Atzori, Fabio Facchinetti
The Journal of Maternal-Fetal & Neonatal Medicine.2024; 35(25): 8275. CrossRef - Branched‐Chain Amino Acid Accumulation Fuels the Senescence‐Associated Secretory Phenotype
Yaosi Liang, Christopher Pan, Tao Yin, Lu Wang, Xia Gao, Ergang Wang, Holly Quang, De Huang, Lianmei Tan, Kun Xiang, Yu Wang, Peter B. Alexander, Qi‐Jing Li, Tso‐Pang Yao, Zhao Zhang, Xiao‐Fan Wang
Advanced Science.2024;[Epub] CrossRef - Exploring the functional roles of small-molecule metabolites in disease research: Recent advancements in metabolomics
Aolei Tan, Xiaoxiao Ma
Chinese Chemical Letters.2024; 35(8): 109276. CrossRef - Deficiency of BCAT2-mediated branched-chain amino acid catabolism promotes colorectal cancer development
Zi-Ran Kang, Shanshan Jiang, Ji-Xuan Han, Yaqi Gao, Yile Xie, Jinxian Chen, Qiang Liu, Jun Yu, Xin Zhao, Jie Hong, Haoyan Chen, Ying-Xuan Chen, Huimin Chen, Jing-Yuan Fang
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2024; 1870(2): 166941. CrossRef - Microbiome metabolite quantification methods enabling insights into human health and disease
Jarrod Roach, Rohit Mital, Jacob J. Haffner, Nathan Colwell, Randy Coats, Horvey M. Palacios, Zongyuan Liu, Joseane L.P. Godinho, Monica Ness, Thilini Peramuna, Laura-Isobel McCall
Methods.2024; 222: 81. CrossRef - Branched-chain amino acids promote occurrence and development of cardiovascular disease dependent on triglyceride metabolism via activation of the mTOR/SREBP-1/betatrophin pathway
Jie Zhang, Ziyu Liu, Yaojun Ni, Yang Yu, Fei Guo, Yanwen Lu, Xiaoqing Wang, Hairong Hao, Shayan Li, Pan Wei, Weinan Yu, Wen Hu
Molecular and Cellular Endocrinology.2024; 584: 112164. CrossRef - Heat stress reduces brown adipose tissue activity by exacerbating mitochondrial damage in type 2 diabetic mice
Penghua Lai, Linlin Zhang, Yan Qiu, Jie Ren, Xue Sun, Ting Zhang, Liuyi Wang, Sijie Cheng, Sijia Liu, Hongli Zhuang, Daiwei Lu, Shaoliang Zhang, Huiqing Liang, Shaodong Chen
Journal of Thermal Biology.2024; 119: 103799. CrossRef - Unraveling Adipose Tissue Dysfunction: Molecular Mechanisms, Novel Biomarkers, and Therapeutic Targets for Liver Fat Deposition
Marta Lopez-Yus, Carlos Hörndler, Sofia Borlan, Vanesa Bernal-Monterde, Jose M. Arbones-Mainar
Cells.2024; 13(5): 380. CrossRef - Microbial production of branched chain amino acids: Advances and perspectives
Yanan Hao, Xuewei Pan, Jiajia You, Guomin Li, Meijuan Xu, Zhiming Rao
Bioresource Technology.2024; 397: 130502. CrossRef - Maintenance of the branched-chain amino acid transporter LAT1 counteracts myotube atrophy following chemotherapy
Stephen Mora, Olasunkanmi A. J. Adegoke
American Journal of Physiology-Cell Physiology.2024; 326(3): C866. CrossRef - Metabolomics of Mice with Type 2 Diabetes and Nonalcoholic Fatty Liver Treated by Acupuncture
Yihui Guo, Liying Zhang, Mengyuan Li, Linan Lin, Fuyu Xue, Wanning Gao, Xiaoru Xu, Haipeng Huang, Abdelilah Arredouani
International Journal of Endocrinology.2024; 2024: 1. CrossRef - Metabolic Signatures Elucidate the Effect of Body Mass Index on Type 2 Diabetes
Qiuling Dong, Sidra Sidra, Christian Gieger, Rui Wang-Sattler, Wolfgang Rathmann, Cornelia Prehn, Jerzy Adamski, Wolfgang Koenig, Annette Peters, Harald Grallert, Sapna Sharma
Metabolites.2023; 13(2): 227. CrossRef - The Preventive Effect of Exercise and Oral Branched-Chain Amino Acid Supplementation on Obesity-Induced Brain Changes in Ldlr−/−.Leiden Mice
Klara J. Lohkamp, Anita M. van den Hoek, Gemma Solé-Guardia, Maria Lisovets, Talissa Alves Hoffmann, Konstantina Velanaki, Bram Geenen, Vivienne Verweij, Martine C. Morrison, Robert Kleemann, Maximilian Wiesmann, Amanda J. Kiliaan
Nutrients.2023; 15(7): 1716. CrossRef - The Crosstalk between Gut Microbiota and White Adipose Tissue Mitochondria in Obesity
Luca Colangeli, David Israel Escobar Marcillo, Valeria Simonelli, Egidio Iorio, Tommaso Rinaldi, Paolo Sbraccia, Paola Fortini, Valeria Guglielmi
Nutrients.2023; 15(7): 1723. CrossRef - Prebiotic and Probiotic Modulation of the Microbiota–Gut–Brain Axis in Depression
Daniel E. Radford-Smith, Daniel C. Anthony
Nutrients.2023; 15(8): 1880. CrossRef - Machine learning model to predict obesity using gut metabolite and brain microstructure data
Vadim Osadchiy, Roshan Bal, Emeran A. Mayer, Rama Kunapuli, Tien Dong, Priten Vora, Danny Petrasek, Cathy Liu, Jean Stains, Arpana Gupta
Scientific Reports.2023;[Epub] CrossRef - Insulin Resistance and Impaired Branched-Chain Amino Acid Metabolism in Alzheimer’s Disease
Rui Liu, Lei Zhang, Hao You
Journal of Alzheimer's Disease.2023; 93(3): 847. CrossRef - Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments
Rexiati Ruze, Tiantong Liu, Xi Zou, Jianlu Song, Yuan Chen, Ruiyuan Xu, Xinpeng Yin, Qiang Xu
Frontiers in Endocrinology.2023;[Epub] CrossRef - Crosstalk between arginine, glutamine, and the branched chain amino acid metabolism in the tumor microenvironment
Tanner J. Wetzel, Sheila C. Erfan, Lucas D. Figueroa, Leighton M. Wheeler, Elitsa A. Ananieva
Frontiers in Oncology.2023;[Epub] CrossRef - Stachydrine, N‐acetylornithine and trimethylamine N‐oxide levels as candidate milk biomarkers of maternal consumption of an obesogenic diet during lactation
Pedro Castillo, Ondrej Kuda, Jan Kopecky, Catalina Amadora Pomar, Andreu Palou, Mariona Palou, Catalina Picó
BioFactors.2023; 49(5): 1022. CrossRef - Biofunctionalization of natural extracts, trends in biological activity and kinetic release
Abraham Osiris Martínez-Olivo, Víctor Manuel Zamora-Gasga, Luis Medina-Torres, Alejandro Pérez-Larios, Sonia Guadalupe Sáyago-Ayerdi, Jorge Alberto Sánchez-Burgos
Advances in Colloid and Interface Science.2023; 318: 102938. CrossRef - Muscle Traits, Sarcopenia, and Sarcopenic Obesity: A Vitamin D Mendelian Randomization Study
Joshua P. Sutherland, Ang Zhou, Elina Hyppönen
Nutrients.2023; 15(12): 2703. CrossRef - Bioactive Ingredients in Traditional Fermented Food Condiments: Emerging Products for Prevention and Treatment of Obesity and Type 2 Diabetes
Alphonse Laya, Honoré Wangso, Iva Fernandes, Raphaël Djakba, Joana Oliveira, Eugenia Carvalho, Wen yi Kang
Journal of Food Quality.2023; 2023: 1. CrossRef - Sex related differences in muscle health and metabolism in chronic obstructive pulmonary disease
Mariëlle P.K.J. Engelen, Sarah K. Kirschner, Kimberly S. Coyle, David Argyelan, Gabriel Neal, Srinivasan Dasarathy, Nicolaas E.P. Deutz
Clinical Nutrition.2023; 42(9): 1737. CrossRef - Depiction of Branched-Chain Amino Acids (BCAAs) in Diabetes with a Focus on Diabetic Microvascular Complications
Daniela Maria Tanase, Evelina Maria Gosav, Tina Botoc, Mariana Floria, Claudia Cristina Tarniceriu, Minela Aida Maranduca, Anca Haisan, Andrei Ionut Cucu, Ciprian Rezus, Claudia Florida Costea
Journal of Clinical Medicine.2023; 12(18): 6053. CrossRef - Genetic and Lifestyle-Related Factors Influencing Serum Hyper-Propionylcarnitine Concentrations and Their Association with Metabolic Syndrome and Cardiovascular Disease Risk
Yong-Hwa Lee, Sunmin Park
International Journal of Molecular Sciences.2023; 24(21): 15810. CrossRef -
Relationship between Components, Intestinal Microbiota, and Mechanism of Hypoglycemic Effect of the Saggy Ink Cap Medicinal Mushroom (Coprinus Comatus, Agaricomycetes): A Review
Wei Wang, Min Sun, Jinyan Yu, Xumin Ma, Chunchao Han
International Journal of Medicinal Mushrooms.2023; 25(12): 81. CrossRef - High-protein diet with excess leucine prevents inactivity-induced insulin resistance in women
Alessandro Mangogna, Filippo Giorgio Di Girolamo, Nicola Fiotti, Pierandrea Vinci, Matteo Landolfo, Filippo Mearelli, Gianni Biolo
Clinical Nutrition.2023; 42(12): 2578. CrossRef - Toward Systems-Level Metabolic Analysis in Endocrine Disorders and Cancer
Aliya Lakhani, Da Hyun Kang, Yea Eun Kang, Junyoung O. Park
Endocrinology and Metabolism.2023; 38(6): 619. CrossRef - Dietary intake of branched-chain amino acids in relation to general and abdominal obesity
Farzaneh Asoudeh, Asma Salari-Moghaddam, Ammar Hassanzadeh Keshteli, Ahmad Esmaillzadeh, Peyman Adibi
Eating and Weight Disorders - Studies on Anorexia, Bulimia and Obesity.2022; 27(4): 1303. CrossRef - Tea polyphenol – gut microbiota interactions: hints on improving the metabolic syndrome in a multi-element and multi-target manner
Hui Ma, Yaozhong Hu, Bowei Zhang, Zeping Shao, Eugeni Roura, Shuo Wang
Food Science and Human Wellness.2022; 11(1): 11. CrossRef - Exogenous isoleucine can confer browning resistance on fresh-cut potato by suppressing polyphenol oxidase activity and improving the antioxidant capacity
Zan Meng, Tong Wang, Aman Ullah Malik, Qingguo Wang
Postharvest Biology and Technology.2022; 184: 111772. CrossRef - Impact of thermal treatment and fermentation by lactic acid bacteria on sorghum metabolite changes, their antioxidant and antidiabetic activities
Fred Kwame Ofosu, Fazle Elahi, Eric Banan-Mwine Daliri, Sang-Ik Han, Deog-Hwan Oh
Food Bioscience.2022; 45: 101502. CrossRef - Infant intakes of human milk branched chain amino acids are negatively associated with infant growth and influenced by maternal body mass index
Jessica L. Saben, Clark R. Sims, Lindsay Pack, Renny Lan, Elisabet Børsheim, Aline Andres
Pediatric Obesity.2022;[Epub] CrossRef - Roux-En-Y Gastric Bypass (RYGB) Surgery during High Liquid Sucrose Diet Leads to Gut Microbiota-Related Systematic Alterations
Laimdota Zizmare, Christina N. Boyle, Sabrina Buss, Sandrine Louis, Laura Kuebler, Ketki Mulay, Ralf Krüger, Lara Steinhauer, Isabelle Mack, Manuel Rodriguez Gomez, Kristina Herfert, Yvonne Ritze, Christoph Trautwein
International Journal of Molecular Sciences.2022; 23(3): 1126. CrossRef - Omics Analyses of Intestinal Microbiota and Hypothalamus Clock Genes in Circadian Disturbance Model Mice Fed with Green Tea Polyphenols
Yuting Zhang, Lu Cheng, Yanan Liu, Ruilin Zhang, Zufang Wu, Kejun Cheng, Xin Zhang
Journal of Agricultural and Food Chemistry.2022; 70(6): 1890. CrossRef - Age, Sex, Body Mass Index, Diet and Menopause Related Metabolites in a Large Homogeneous Alpine Cohort
Vinicius Verri Hernandes, Nikola Dordevic, Essi Marjatta Hantikainen, Baldur Bragi Sigurdsson, Sigurður Vidir Smárason, Vanessa Garcia-Larsen, Martin Gögele, Giulia Caprioli, Ilaria Bozzolan, Peter P. Pramstaller, Johannes Rainer
Metabolites.2022; 12(3): 205. CrossRef - α-ketoisocaproic acid promotes ER stress through impairment of autophagy, thereby provoking lipid accumulation and insulin resistance in murine preadipocytes
Tae Jun Park, Seung Yeon Park, Hyun Jung Lee, A.M. Abd El-Aty, Ji Hoon Jeong, Tae Woo Jung
Biochemical and Biophysical Research Communications.2022; 603: 109. CrossRef - The associations of serum valine with mild cognitive impairment and Alzheimer’s disease
Yong-lan Xiong, Joseph Therriault, Shu-jiang Ren, Xiao-jun Jing, Hua Zhang
Aging Clinical and Experimental Research.2022; 34(8): 1807. CrossRef - The Critical Role of the Branched Chain Amino Acids (BCAAs) Catabolism-Regulating Enzymes, Branched-Chain Aminotransferase (BCAT) and Branched-Chain α-Keto Acid Dehydrogenase (BCKD), in Human Pathophysiology
Aikaterini Dimou, Vasilis Tsimihodimos, Eleni Bairaktari
International Journal of Molecular Sciences.2022; 23(7): 4022. CrossRef - Obesity, exercise training, and BCAA supplementation: All that glitters (may not be) gold
Stephen J. Carter, Emily B. Long, Cydne A. Perry
Obesity.2022; 30(6): 1139. CrossRef - Effects of serum branched-chain amino acids on nonalcoholic fatty liver disease and subsequent cardiovascular disease
Fei Guo, Rui Chen, Linghui Kong, Pan Wei, Ziyu Liu, Xiaoqing Wang, Hairong Hao, Yanwen Lu, Wen Hu
Hepatology International.2022; 16(6): 1424. CrossRef - Advances in multi-omics study of biomarkers of glycolipid metabolism disorder
Xinyi Fang, Runyu Miao, Jiahua Wei, Haoran Wu, Jiaxing Tian
Computational and Structural Biotechnology Journal.2022; 20: 5935. CrossRef - Targeted Metabolomics Revealed a Sex-Dependent Signature for Metabolic Syndrome in the Mexican Population
Berenice Palacios-González, Guadalupe León-Reyes, Berenice Rivera-Paredez, Isabel Ibarra-González, Marcela Vela-Amieva, Yvonne N. Flores, Samuel Canizales-Quinteros, Jorge Salmerón, Rafael Velázquez-Cruz
Nutrients.2022; 14(18): 3678. CrossRef - Case report: NAFLD and maple syrup urine disease: Is there an interplay between branched-chain amino acids and fructose consumption?
Helena Moreira-Silva, Sandra Ferreira, Manuela Almeida, Isabel Gonçalves, Maria Augusta Cipriano, J. R. Vizcaíno, Ermelinda Santos-Silva, Esmeralda Gomes-Martins
Frontiers in Pediatrics.2022;[Epub] CrossRef - A self-assembled leucine polymer sensitizes leukemic stem cells to chemotherapy by inhibiting autophagy in acute myeloid leukemia
Xi Xu, Jian Wang, Tong Tong, Wenwen Zhang, Jin Wang, Weiwei Ma, Shunqing Wang, Dunhua Zhou, Jun Wu, Linjia Jiang, Meng Zhao
Haematologica.2022; 107(10): 2344. CrossRef - Effect of dietary protein content shift on aging in elderly rats by comprehensive quantitative score and metabolomics analysis
Wenxuan Zheng, Ruiding Li, Yang Zhou, Fengcui Shi, Yao Song, Yanting Liao, Fan Zhou, Xiaohua Zheng, Jingwen Lv, Quanyang Li
Frontiers in Nutrition.2022;[Epub] CrossRef - Caloric restriction improves glycaemic control without reducing plasma branched-chain amino acids or keto-acids in obese men
M. H. Sayda, M. H. Abdul Aziz, N. Gharahdaghi, D. J. Wilkinson, P. L. Greenhaff, B. E. Phillips, K. Smith, I. Idris, P. J. Atherton
Scientific Reports.2022;[Epub] CrossRef - Dietary Protein and Amino Acid Deficiency Inhibit Pancreatic Digestive Enzyme mRNA Translation by Multiple Mechanisms
Maria Dolors Sans, Stephen J. Crozier, Nancy L. Vogel, Louis G. D’Alecy, John A. Williams
Cellular and Molecular Gastroenterology and Hepatology.2021; 11(1): 99. CrossRef - Isoleucine increases muscle mass through promoting myogenesis and intramyocellular fat deposition
Shuge Liu, Yunmei Sun, Rui Zhao, Yingqian Wang, Wanrong Zhang, Weijun Pang
Food & Function.2021; 12(1): 144. CrossRef - Serum metabolomics study of women with different annual decline rates of anti-Müllerian hormone: an untargeted gas chromatography–mass spectrometry-based study
Nazanin Moslehi, Parvin Mirmiran, Rezvan Marzbani, Hassan Rezadoost, Mehdi Mirzaie, Fereidoun Azizi, Fahimeh Ramezani Tehrani
Human Reproduction.2021; 36(3): 721. CrossRef - Amino acid sensing pathway: A major check point in the pathogenesis of obesity and COVID‐19
Aradhana Mariam Philips, Nooruddin Khan
Obesity Reviews.2021;[Epub] CrossRef - The chemical mechanisms of the enzymes in the branched-chain amino acids biosynthetic pathway and their applications
Yan-Fei Liang, Zi-Xian Long, Ya-Jian Zhang, Cai-Yun Luo, Le-Tian Yan, Wen-Yun Gao, Heng Li
Biochimie.2021; 184: 72. CrossRef - Plasma Metabolomic Profiling in 1391 Subjects with Overweight and Obesity from the SPHERE Study
Gianfranco Frigerio, Chiara Favero, Diego Savino, Rosa Mercadante, Benedetta Albetti, Laura Dioni, Luisella Vigna, Valentina Bollati, Angela Cecilia Pesatori, Silvia Fustinoni
Metabolites.2021; 11(4): 194. CrossRef - Sarcopenic Obesity and Amino Acids: Concord Health and Ageing in Men Project
David G Le Couteur, David J Handelsman, Fiona Stanaway, Louise M Waite, Fiona M Blyth, Vasi Naganathan, Robert G Cumming, Vasant Hirani, Rafael de Cabo
The Journals of Gerontology: Series A.2021; 76(6): 1000. CrossRef - Metabolic signatures of greater body size and their associations with risk of colorectal and endometrial cancers in the European Prospective Investigation into Cancer and Nutrition
Nathalie Kliemann, Vivian Viallon, Neil Murphy, Rebecca J. Beeken, Joseph A. Rothwell, Sabina Rinaldi, Nada Assi, Eline H. van Roekel, Julie A. Schmidt, Kristin Benjaminsen Borch, Claudia Agnoli, Ann H. Rosendahl, Hanna Sartor, José María Huerta, Anne Tjø
BMC Medicine.2021;[Epub] CrossRef - Plasma Amino Acids and Residual Hypertriglyceridemia in Diabetic Patients Under Statins: Two Independent Cross-Sectional Hospital-Based Cohorts
Shuang Wang, Yun-Feng Cao, Xiao-Yu Sun, Mo Hong, Zhong-Ze Fang, Hui-Huan Luo, Huan Sun, Ping Yang
Frontiers in Cardiovascular Medicine.2021;[Epub] CrossRef - Identification of Metabolic Phenotypes in Young Adults with Obesity by 1H NMR Metabolomics of Blood Serum
Khin Thandar Htun, Jie Pan, Duanghathai Pasanta, Montree Tungjai, Chatchanok Udomtanakunchai, Sirirat Chancharunee, Siriprapa Kaewjaeng, Hong Joo Kim, Jakrapong Kaewkhao, Suchart Kothan
Life.2021; 11(6): 574. CrossRef - A Metabolic Pattern in Healthy Subjects Given a Single Dose of Metformin: A Metabolomics Approach
Lina A. Dahabiyeh, Muhammad Mujammami, Tawfiq Arafat, Hicham Benabdelkamel, Assim A. Alfadda, Anas M. Abdel Rahman
Frontiers in Pharmacology.2021;[Epub] CrossRef - Branched-chain Amino Acids: Catabolism in Skeletal Muscle and Implications for Muscle and Whole-body Metabolism
Gagandeep Mann, Stephen Mora, Glory Madu, Olasunkanmi A. J. Adegoke
Frontiers in Physiology.2021;[Epub] CrossRef - Identification of TBX15 as an adipose master trans regulator of abdominal obesity genes
David Z. Pan, Zong Miao, Caroline Comenho, Sandhya Rajkumar, Amogha Koka, Seung Hyuk T. Lee, Marcus Alvarez, Dorota Kaminska, Arthur Ko, Janet S. Sinsheimer, Karen L. Mohlke, Nicholas Mancuso, Linda Liliana Muñoz-Hernandez, Miguel Herrera-Hernandez, Maria
Genome Medicine.2021;[Epub] CrossRef - Antidiabetic Effect of Noodles Containing Fermented Lettuce Extracts
Soon Yeon Jeong, Eunjin Kim, Ming Zhang, Yun-Seong Lee, Byeongjun Ji, Sun-Hee Lee, Yu Eun Cheong, Soon-Il Yun, Young-Soo Kim, Kyoung Heon Kim, Min Sun Kim, Hyun Soo Chun, Sooah Kim
Metabolites.2021; 11(8): 520. CrossRef - Targeted and Untargeted Mass Spectrometry Reveals the Impact of High-Fat Diet on Peripheral Amino Acid Regulation in a Mouse Model of Alzheimer’s Disease
Amelia L. Taylor, Don E. Davis, Simona G. Codreanu, Fiona E. Harrison, Stacy D. Sherrod, John A. McLean
Journal of Proteome Research.2021; 20(9): 4405. CrossRef - The Association of 9 Amino Acids With Cardiovascular Events in Finnish Men in a 12-Year Follow-up Study
Raimo Jauhiainen, Jagadish Vangipurapu, Annamaria Laakso, Teemu Kuulasmaa, Johanna Kuusisto, Markku Laakso
The Journal of Clinical Endocrinology & Metabolism.2021; 106(12): 3448. CrossRef - Serum Metabolite Profile Associated with Sex-Dependent Visceral Adiposity Index and Low Bone Mineral Density in a Mexican Population
Berenice Palacios-González, Guadalupe León-Reyes, Berenice Rivera-Paredez, Isabel Ibarra-González, Marcela Vela-Amieva, Yvonne N. Flores, Samuel Canizales-Quinteros, Jorge Salmerón, Rafael Velázquez-Cruz
Metabolites.2021; 11(9): 604. CrossRef - Branched chain amino acids—friend or foe in the control of energy substrate turnover and insulin sensitivity?
Elżbieta Supruniuk, Ewa Żebrowska, Adrian Chabowski
Critical Reviews in Food Science and Nutrition.2021; : 1. CrossRef - Diet Effects on Cerebrospinal Fluid Amino Acids Levels in Adults with Normal Cognition and Mild Cognitive Impairment
Kate J. Russin, K. Sreekumaran Nair, Thomas J. Montine, Laura D. Baker, Suzanne Craft
Journal of Alzheimer's Disease.2021; 84(2): 843. CrossRef - QSHY Granules Promote White Adipose Tissue Browning and Correct BCAAs Metabolic Disorder in NAFLD Mice
Binbin Zhang, Mingzhu Ni, Xiaojing Li, Qiaohong Liu, Yiyang Hu, Yu Zhao
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy.2021; Volume 14: 4241. CrossRef - The Changes in Endogenous Metabolites in Hyperlipidemic Rats Treated with Herbal Mixture Containing Lemon, Apple Cider, Garlic, Ginger, and Honey
Azliana Abu Bakar Sajak, Azrina Azlan, Faridah Abas, Hazilawati Hamzah
Nutrients.2021; 13(10): 3573. CrossRef - Integration of Transcriptome and Metabolome Provides Unique Insights to Pathways Associated With Obese Breast Cancer Patients
Mohammed A. Hassan, Kaltoom Al-Sakkaf, Mohammed Razeeth Shait Mohammed, Ashraf Dallol, Jaudah Al-Maghrabi, Alia Aldahlawi, Sawsan Ashoor, Mabrouka Maamra, Jiannis Ragoussis, Wei Wu, Mohammad Imran Khan, Abdulrahman L. Al-Malki, Hani Choudhry
Frontiers in Oncology.2020;[Epub] CrossRef - Omics Biomarkers in Obesity: Novel Etiological Insights and Targets for Precision Prevention
Krasimira Aleksandrova, Caue Egea Rodrigues, Anna Floegel, Wolfgang Ahrens
Current Obesity Reports.2020; 9(3): 219. CrossRef - Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes?
Milan Holeček
Nutrients.2020; 12(10): 3087. CrossRef - Differential gene signature in adipose tissue depots of growth hormone transgenic mice
Silvana Duran‐Ortiz, Jonathan A. Young, Adam Jara, Elizabeth A. Jensen, Reetobrata Basu, Edward O. List, Yanrong Qian, John J. Kopchick, Darlene E. Berryman
Journal of Neuroendocrinology.2020;[Epub] CrossRef - Prognostic Role of Serum Amino Acids in Head and Neck Cancer
Gabriella Cadoni, Luca Giraldi, Carlo Chiarla, Jacopo Gervasoni, Silvia Persichilli, Aniello Primiano, Stefano Settimi, Jacopo Galli, Gaetano Paludetti, Dario Arzani, Stefania Boccia, Ivo Giovannini, Giovanni Almadori, Leigh A. Madden
Disease Markers.2020; 2020: 1. CrossRef - Lipid changes in the metabolome of a single case study with maple syrup urine disease (MSUD) after five days of improved diet adherence of controlled branched-chain amino acids (BCAA)
Teresa D. Douglas, L. Kristin Newby, Julie Eckstrand, Douglas Wixted, Rani H. Singh
Molecular Genetics and Metabolism Reports.2020; 25: 100651. CrossRef - Branched chain amino acids, aging and age-related health
David G. Le Couteur, Samantha M. Solon-Biet, Victoria C. Cogger, Rosilene Ribeiro, Rafael de Cabo, David Raubenheimer, Gregory J. Cooney, Stephen J. Simpson
Ageing Research Reviews.2020; 64: 101198. CrossRef - Branched-chain ketoacid overload inhibits insulin action in the muscle
Dipsikha Biswas, Khoi T. Dao, Angella Mercer, Andrew M. Cowie, Luke Duffley, Yassine El Hiani, Petra C. Kienesberger, Thomas Pulinilkunnil
Journal of Biological Chemistry.2020; 295(46): 15597. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

