
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 31(2); 2016 > Article
-
Review ArticleHigh-Density Lipoprotein, Lecithin: Cholesterol Acyltransferase, and Atherosclerosis
-
Alice Ossoli, Chiara Pavanello, Laura Calabresi

-
Endocrinology and Metabolism 2016;31(2):223-229.
DOI: https://doi.org/10.3803/EnM.2016.31.2.223
Published online: June 10, 2016
Center E. Grossi Paoletti, Department of Pharmacological and Biomolecular Sciences, University of Milano, Milano, Italy.
- Corresponding author: Laura Calabresi. Center E. Grossi Paoletti, Department of Pharmacological and Biomolecular Sciences, University of Milano, Via Balzaretti 9, Milano 20133, Italy. Tel: +39-250319906, Fax: +39-25031900, laura.calabresi@unimi.it
• Received: May 3, 2016 • Revised: May 11, 2016 • Accepted: May 18, 2016
Copyright © 2016 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Epidemiological data clearly show the existence of a strong inverse correlation between plasma high-density lipoprotein cholesterol (HDL-C) concentrations and the incidence of coronary heart disease. This relation is explained by a number of atheroprotective properties of HDL, first of all the ability to promote macrophage cholesterol transport. HDL are highly heterogeneous and are continuously remodeled in plasma thanks to the action of a number of proteins and enzymes. Among them, lecithin:cholesterol acyltransferase (LCAT) plays a crucial role, being the only enzyme able to esterify cholesterol within lipoproteins. LCAT is synthetized by the liver and it has been thought to play a major role in reverse cholesterol transport and in atheroprotection. However, data from animal studies, as well as human studies, have shown contradictory results. Increased LCAT concentrations are associated with increased HDL-C levels but not necessarily with atheroprotection. On the other side, decreased LCAT concentration and activity are associated with decreased HDL-C levels but not with increased atherosclerosis. These contradictory results confirm that HDL-C levels per se do not represent the functionality of the HDL system.
- A large number of studies has clearly shown the existence of a strong inverse correlation between plasma high-density lipoprotein cholesterol (HDL-C) concentrations and the incidence of coronary heart disease (CHD) [12], but the significance of such association has been recently questioned. Intervention clinical trials carried out with agents efficient in raising HDL-C levels, including niacin and cholesteryl ester transfer protein (CETP) inhibitors, have failed in showing a reduction in cardiovascular events [345]. In addition, Mendelian randomization studies have shown that increased HDL-C levels caused by common variants in HDL related genes are not necessarily associated with reduced cardiovascular risk [678]. One possible explanation for this discrepancy is that the plasma HDL-C concentration does not reflect the very complex HDL system, involving different HDL particles and a number of receptors, transporters, enzyme, and transfer proteins [9]. Moreover, cholesterol is not the active component of HDL and there is convincing evidence that at least some of the atheroprotective functions of HDL relate to specific HDL components or subclasses, which concentration in plasma may be totally unrelated to the HDL-C level [1011].
INTRODUCTION
- HDL heterogeneity
- HDL are a highly heterogeneous lipoprotein family composed by several subclasses with different density, shape, and size. The density of the HDL particles is inversely related to their size, reflecting the relative contents of low density non-polar core lipid, and high density surface protein. Most part of plasma HDL has a globular shape, the central core is composed by non-polar lipids (triglycerides and cholesteryl esters) surrounded by a monolayer of polar lipids (phospholipids and unesterified cholesterol) and apolipoproteins [12]. A minor fraction of plasma HDL has a non-spherical structure and they consist in discoidal bilayer of polar lipids, in which non-polar core is lacking; apolipoproteins run from side to side of the disk, with polar residues facing the aqueous phase and non-polar residues facing the acyl chains of the lipid bilayer [13]. The protein component of HDL is formed mainly by apolipoprotein A-I (apoA-I), for the 70%, and apolipoprotein A-II (apoA-II), for the 20%. Two major particle subclasses have been identified on the basis of major apolipoprotein composition: particles containing only apoA-I (LpA-I), and particles containing both apoA-I and apoA-II (LpA-I:A-II). Recent shotgun proteomic analysis showed that HDL contain 48 or more proteins, among these apoA-IV, apoCs, apoE, lecithin:cholesterol acyltransferase (LCAT), CETP, phospholipid transfer protein (PLTP), paraoxonase (PON), and platelet-activating factor acetylhydrolase (PAF-AH) circulate in plasma bound to HDL [91214]. Most of the proteins carried by HDL are not apolipoproteins, and represent very minor components of these particles.
- It is also possible to classify HDL on the basis of density (HDL2, with density of 1.063 to 1.120 g/mL, and HDL3, 1.120 to 1.210 g/mL), charge, shape, and size. According to charge, HDL can be divided into α- and pre-β-migrating particles on agarose gel, and combining charge and size these two subclass-es can be divided into 12 distinct apoA-I-containing particles, referred to as pre-β (pre-β1 and pre-β2), α (α1, α2, and α3) and pre-α (pre-α1, pre-α2, and pre-α3) on the basis of mobility that is slower or faster than albumin, respectively, and decreasing size [15].
- HDL metabolism
- Due to its highly dynamic nature, apoA-I is involved in major pathways of HDL metabolism. ApoA-I and apoA-II are synthe-sized mainly by the liver and, to a lesser extent, by the small intestine [16] and are secreted as components of triglyceride-rich lipoproteins. In circulation, PLTP promotes the transfer of surface components (phospholipids, cholesterol, and apolipoproteins) from triglyceride-rich lipoproteins to HDL. The regulatory role of PLTP is achieved through two main functions, phospholipid transfer activity and the capability to modulate HDL size and composition in a process called HDL conversion [17]. Hepatocytes are able to secrete apoA-I in lipid-free or lipid-poor and lipidated forms [18]. ApoA-I is secreted as pro-apoA-I and converted to a mature form by a metalloprotease in plasma [19]. There are three potential sources of lipid-poor apoA-I in plasma: it may be released as lipid-poor protein after its synthesis in the liver and intestine, it may be released from triglyceride-rich lipoproteins that are undergoing lipolysis by lipoprotein lipase, and it may be generated in the circulation during the remodeling of mature, spherical HDL particles. Lipid free-apoA-I acquires phospholipids and cholesterol through the interaction with the ATP binding cassette transporter A1 (ABCA1), to form pre-β-HDL, a pathway dependent on ABCA1 expression [20]. Once in the circulation, pre-β-HDL are the preferential substrate of LCAT (Fig. 1), that converts lecithin and cholesterol into lysolecithin and cholesteryl esters, using apoA-I as cofactor [21]. The cholesterol esters generated by LCAT are more hydrophobic than free cholesterol and thus migrate into the hydrophobic core of the lipoprotein, with the resulting conversion of small, discoidal pre-β-HDL into mature, spherical, α-migrating HDL (α-HDL) [21]. LCAT thus plays a central role in intravascular HDL metabolism and in the determination of plasma HDL level. Esterification of cholesterol in plasma by LCAT is also necessary for cholesterol uptake from the liver, either directly through the scavenger receptor class B member 1 (SR-BI) or indirectly through CETP. The α-HDL produced by LCAT (HDL3) interact in the plasma with CETP, that exchanges cholesteryl esters for triglycerides between HDL and triglyceride-rich lipoproteins, generating large cholesteryl ester-poor and triglyceride-rich HDL particles (HDL2) [22]. Mature, large α-HDL particles can be converted back to pre-β-HDL through the action of PLTP and the endothelial and hepatic lipases, that hydrolyze triglycerides and phospholipids on HDL (Fig. 1) [23]. Plasma half-life of pre-β-HDL is short, they are rapidly cleared through the kidney, while mature α-HDL have a slower turnover [24].
- HDL components are catabolized in different ways; the major sites of catabolism of the protein components are liver and kidney. The kidney, according to hydrophobicity, filters lipid-free apolipoproteins; apoA-I and apoA-II can be reabsorbed through cubulin receptors in the kidney proximal tubules [25]. When reabsorption is impaired, hydrophilic apolipoproteins (apoA-I and apoA-IV, but no apoA-II) can be excreted into urine [26]. Glomerular filtration barrier prevents access of mature HDL particles to the proximal tubules; however, cubulin may bind filtered lipid-poor HDL [27]. HDL particles can entirely be removed by holoparticles HDL receptors. In the liver, holo-HDL particles accumulated in endosomal compartments can be transferred to lysosomes for degradation or in a small proportion, they can be resecreted into the circulation [28].
- HDL functions
- One of the most important function of HDL is to promote the removal of cholesterol from peripheral cells, including macrophages within the arterial wall, and shuttle it to the liver for excretion through the bile and feces in a process called reverse cholesterol transport (RCT). It results in a net mass transport of cholesterol from the arterial wall into the bile. This pathway is described as an anti-atherogenic process by preventing arterial cholesterol accumulation, plaque destabilization, and development of acute cardiovascular events [9]. Cell cholesterol efflux is the first and limiting step in RCT and consists in the exchange of unesterified cholesterol between cells and extracellular acceptors [29]. This exchange can occur by several processes: via aqueous diffusion, which occurs according to the direction of cholesterol gradient, or through three main distinct and protein-mediated pathways [29]. The various HDL particles are differently efficient in promoting cell cholesterol efflux. Lipid-free/lipid-poor apolipoproteins, mainly apoA-I, represent the principal cholesterol acceptors via ABCA1 [29]. All plasma HDL subclasses, including mature α-HDL particles and discoidal pre-β-HDL, are efficient cholesterol acceptors via the ABCG1 pathway, while SR-BI promotes cell cholesterol efflux only to mature, large α-HDL [30]. The cholesterol accumulated in HDL is esterified in plasma by LCAT with the resulting formation of cholesteryl esters. Then, hydrophobic cholesteryl esters move to the core while unesterified cholesterol is removed from the surface of HDL, leading to the progressive enlargement of these particles. For long time, LCAT has been considered necessary for efficient RCT by keeping unesterified cholesterol gradient from cells to HDL, but recent data suggest that even if functional LCAT is not present, macrophage cholesterol efflux and RCT can occur [31]. Large amount of the cholesteryl esters formed by the LCAT are exchanged with triglycerides through CETP-mediated process into apoB containing lipoproteins that are finally catabolized by the liver. Alternatively, HDL-cholesteryl esters are taken-up by the liver through SR-BI [32].
- Atheroprotection mediated by HDL is not only through their major role in RCT, but also through other relevant functions; one of the most important and well studied is the ability of HDL to maintain endothelial cell homeostasis and integrity [33]. HDL have potent antioxidant properties, mediated by molecules carried by HDL (PON-1, PAF-AH, and LCAT) or by apoA-I and apoA-II, as well as anti-inflammatory, antithrombotic, cytoprotective, vasodilatory, anti-infectious activities and the capacity to enhance insulin secretion. This wide spectrum of biological activities likely reflects the heterogeneity of HDL particles; however, the HDL-protective activities can be lost in some pathological conditions and HDL can even acquire proatherogenic properties [34].
HDL METABOLISM AND FUNCTION
- LCAT is principally synthetized in the liver and in little amount in other tissues, such as brain and testes, and circulates in plasma compartment at concentration of 5 mg/L mainly bound to HDL but also to LDL. LCAT converts phosphatidylcholine and cholesterol into cholesteryl ester and lysophosphatidylcholine in plasma and other biological fluids [21]. In RCT pathway, LCAT plays a key role and it is thought to help facilitate this process by leading formation of large and mature HDL. Furthermore, it is reported that the majority of cholesteryl esters formed by LCAT are removed by the liver [35]. Without LCAT, HDL-C, apoA-I and apoA-II levels in the plasma are drastically reduced for the lacking formation of mature and spherical shaped HDL and for the rapid catabolism of discoidal HDL by the kidney [36]. On the basis of these evidences, variations in LCAT activity seem to be naturally implicated in atherosclerosis prevention or development. To elucidate the role of LCAT in atherosclerosis, a large number of studies have been performed in both animal models and humans (Table 1) [837383940414243444546474849505152].
- Studies carried out in animal models led to controversial results, often dependent on species utilized. Studies performed in mice in which LCAT was overexpressed [373839] or downregulated [4041] suggest that activity of LCAT is not associated to atheroprotection and the lack of enzyme is not associated to increased atherosclerosis, even if the HDL-C levels in plasma are very low. The increased atherosclerosis in mice with LCAT overexpression is probably due to the accumulation in plasma of dysfunctional large apoE-rich HDL, which were shown to be defective in the delivery of cholesterol to the liver through SR-BI [39]. When the LCAT gene was overexpressed in rabbits, opposite results were obtained: aortic lesions were reduced after atherogenic diet, even if large HDL particles containing apoE were detected [4243]. The contradictory results obtained in the studies on animal models do not clarify the role of LCAT in atherosclerosis, allowing for further consideration.
- The role of LCAT in atherosclerosis was also explored in humans, both in general population and in subjects at high cardiovascular risk. As observed in animal studies, the role of LCAT in the pathogenesis of human atherosclerosis remains controversial. The Epic-Norfolk was the first prospective study investigating the correlation of LCAT plasma levels and atherosclerosis carried out in general population in more than 2,700 subjects [44]. One-third of enrolled subjects developed coronary artery diseases (CADs), but no associations between plasma LCAT levels and risk to develop future CAD was observed. When individuals were divided according to gender, increased LCAT levels correlated with lower risk of CAD only in men, while in women was the opposite [44]. Reduction of LCAT concentration/activity associated with absence of CAD was described in The Copenhagen City Heart Study, that enrolled more than 10,000 participants, and in The Copenhagen General Population Study, in which more than 50,000 subjects are involved [8]. The variants S208T found in the coding region of LCAT gene was associated with reduction in HDL-C and apoA-I levels, but not with increased risk of myocardial infarction, ischemic heart disease, and ischemic cerebrovascular disease [8]. In agreement with the results obtained in the general population, an observational study carried out in 540 subjects at high cardiovascular risk showed that low plasma LCAT levels are not associated with higher carotid intima-media thickness (IMT) [45], a marker of preclinical atherosclerosis.
- Consistent with these results, in various studies it was demonstrated that an increased LCAT concentration is associated to CAD. Increased levels of LCAT activity was associated with increased IMT in 74 subjects with metabolic syndrome [46], as well as in the control subjects of the study [46]. In another study from the same group, the association between LCAT activity and CAD was found only in men [47]. A recent study analyzed the relationship between LCAT activity and triglyceride metabolism and LDL particle size in 550 patients at high cardiovascular risk [48]. Increased LCAT activity was associated with formation of small LDL particles that are more atherogenic than large particles, but no parameters of subclinical atherosclerosis were analyzed [48].
- On other side, some studies affirm the opposite: decreased LCAT activity is associated with CAD. Early studies supporting this evidence were carried out in 1973 in subjects at high cardiovascular risk [49]. Few years later, in 100 subjects divided according to the degree of atherosclerotic disease, LCAT activity was found positively correlated with the severity of coronary atherosclerosis [50]. Lower levels of LCAT activity were also observed in patients with ischemic heart disease [51], and in a study on patients with acute myocardial infarction [52].
LCAT AND ATHEROSCLEROSIS
- While epidemiological studies have repeatedly shown a strong and inverse correlation between plasma HDL-C concentrations and the incidence of CHD, the significance of such association for CHD development has been recently questioned, and clinical trials with various drugs able to increase HDL-C levels did not show the expected benefits. HDL metabolism is regulated by a large number of factors that modify plasma levels of circulating HDL, and plasma HDL-C levels are remarkably susceptible to variations in these factors which also affect HDL shape, size, density, and lipid and apolipoprotein composition, and as a consequence HDL function. Investigations of factors involved in HDL metabolism thus represent a good way to understand the relationship between HDL and CHD, and will likely translate in the development of innovative therapeutic approaches to CHD prevention and treatment specifically affecting HDL function independent of plasma HDL-C levels.
CONCLUSIONS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 1977;62:707–714. ArticlePubMed
- 2. Assmann G, Schulte H, von Eckardstein A, Huang Y. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 1996;124(Suppl):S11–S20. ArticlePubMed
- 3. Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007;357:2109–2122. ArticlePubMed
- 4. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 2012;367:2089–2099. ArticlePubMed
- 5. AIM-HIGH Investigators. Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011;365:2255–2267. ArticlePubMed
- 6. Johannsen TH, Kamstrup PR, Andersen RV, Jensen GB, Sillesen H, Tybjaerg-Hansen A, et al. Hepatic lipase, genetically elevated high-density lipoprotein, and risk of ischemic cardiovascular disease. J Clin Endocrinol Metab 2009;94:1264–1273. ArticlePubMedPDF
- 7. Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012;380:572–580. ArticlePubMedPMC
- 8. Haase CL, Tybjærg-Hansen A, Qayyum AA, Schou J, Nordestgaard BG, Frikke-Schmidt R. LCAT, HDL cholesterol and ischemic cardiovascular disease: a Mendelian randomization study of HDL cholesterol in 54,500 individuals. J Clin Endocrinol Metab 2012;97:E248–E256. ArticlePubMed
- 9. Calabresi L, Gomaraschi M, Franceschini G. High-density lipoprotein quantity or quality for cardiovascular prevention? Curr Pharm Des 2010;16:1494–1503. ArticlePubMed
- 10. Sirtori CR, Calabresi L, Franceschini G, Baldassarre D, Amato M, Johansson J, et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: the Limone sul Garda study. Circulation 2001;103:1949–1954. ArticlePubMed
- 11. Rader DJ. Lecithin:cholesterol acyltransferase and atherosclerosis: another high-density lipoprotein story that doesn't quite follow the script. Circulation 2009;120:549–552. ArticlePubMedPMC
- 12. Heinecke JW. The HDL proteome: a marker and perhaps mediator of coronary artery disease. J Lipid Res 2009;50(Suppl):S167–S171. ArticlePubMedPMC
- 13. Gu F, Jones MK, Chen J, Patterson JC, Catte A, Jerome WG, et al. Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J Biol Chem 2010;285:4652–4665. ArticlePubMed
- 14. Tall AR, Jiang Xc, Luo Y, Silver D. 1999 George Lyman Duff memorial lecture: lipid transfer proteins, HDL metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol 2000;20:1185–1188. ArticlePubMed
- 15. Asztalos BF, Schaefer EJ. High-density lipoprotein subpopulations in pathologic conditions. Am J Cardiol 2003;91(7A):12E–17E. ArticlePubMed
- 16. Ikewaki K, Zech LA, Brewer HB Jr, Rader DJ. ApoA-II kinetics in humans using endogenous labeling with stable isotopes: slower turnover of apoA-II compared with the exogenous radiotracer method. J Lipid Res 1996;37:399–407. ArticlePubMed
- 17. Huuskonen J, Olkkonen VM, Jauhiainen M, Ehnholm C. The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis 2001;155:269–281. ArticlePubMed
- 18. Chisholm JW, Burleson ER, Shelness GS, Parks JS. ApoA-I secretion from HepG2 cells: evidence for the secretion of both lipid-poor apoA-I and intracellularly assembled nascent HDL. J Lipid Res 2002;43:36–44. ArticlePubMed
- 19. Edelstein C, Gordon JI, Toscas K, Sims HF, Strauss AW, Scanu AM. In vitro conversion of proapoprotein A-I to apoprotein A-I. Partial characterization of an extracellular enzyme activity. J Biol Chem 1983;258:11430–11433. ArticlePubMed
- 20. Tsujita M, Wu CA, Abe-Dohmae S, Usui S, Okazaki M, Yokoyama S. On the hepatic mechanism of HDL assembly by the ABCA1/apoA-I pathway. J Lipid Res 2005;46:154–162. ArticlePubMed
- 21. Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta 2000;1529:245–256. ArticlePubMed
- 22. Zechner R, Dieplinger H, Steyrer E, Groener J, Calvert D, Kostner GM. In vitro formation of HDL-2 from HDL-3 and triacylglycerol-rich lipoproteins by the action of lecithin: cholesterol acyltransferase and cholesterol ester transfer protein. Biochim Biophys Acta 1987;918:27–35. ArticlePubMed
- 23. Clay MA, Newnham HH, Forte TM, Barter PI. Cholesteryl ester transfer protein and hepatic lipase activity promote shedding of apo A-I from HDL and subsequent formation of discoidal HDL. Biochim Biophys Acta 1992;1124:52–58. ArticlePubMed
- 24. Rye KA, Barter PJ. Formation and metabolism of prebetamigrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol 2004;24:421–428. ArticlePubMed
- 25. Moestrup SK, Kozyraki R. Cubilin, a high-density lipoprotein receptor. Curr Opin Lipidol 2000;11:133–140. ArticlePubMed
- 26. Graversen JH, Castro G, Kandoussi A, Nielsen H, Christensen EI, Norden A, et al. A pivotal role of the human kidney in catabolism of HDL protein components apolipoprotein A-I and A-IV but not of A-II. Lipids 2008;43:467–470. ArticlePubMed
- 27. Moestrup SK, Nielsen LB. The role of the kidney in lipid metabolism. Curr Opin Lipidol 2005;16:301–306. ArticlePubMed
- 28. Rohrl C, Stangl H. HDL endocytosis and resecretion. Biochim Biophys Acta 2013;1831:1626–1633. ArticlePubMedPMC
- 29. Jessup W, Gelissen IC, Gaus K, Kritharides L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol 2006;17:247–257. ArticlePubMed
- 30. Favari E, Calabresi L, Adorni MP, Jessup W, Simonelli S, Franceschini G, et al. Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 2009;48:11067–11074. ArticlePubMed
- 31. Tanigawa H, Billheimer JT, Tohyama J, Fuki IV, Ng DS, Rothblat GH, et al. Lecithin:cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 2009;120:160–169. ArticlePubMedPMC
- 32. Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL, et al. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem 1999;274:33398–33402. ArticlePubMed
- 33. Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol 2003;23:1724–1731. ArticlePubMed
- 34. Kontush A. HDL-mediated mechanisms of protection in cardiovascular disease. Cardiovasc Res 2014;103:341–349. ArticlePubMedPDF
- 35. Turner S, Voogt J, Davidson M, Glass A, Killion S, Decaris J, et al. Measurement of reverse cholesterol transport pathways in humans: in vivo rates of free cholesterol efflux, esterification, and excretion. J Am Heart Assoc 2012;1:e001826ArticlePubMedPMC
- 36. Horowitz BS, Goldberg IJ, Merab J, Vanni TM, Ramakrishnan R, Ginsberg HN. Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J Clin Invest 1993;91:1743–1752. ArticlePubMedPMC
- 37. Vaisman BL, Klein HG, Rouis M, Berard AM, Kindt MR, Talley GD, et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem 1995;270:12269–12275. ArticlePubMed
- 38. Mehlum A, Gjernes E, Solberg LA, Hagve TA, Prydz H. Overexpression of human lecithin:cholesterol acyltransferase in mice offers no protection against diet-induced atherosclerosis. APMIS 2000;108:336–342. ArticlePubMed
- 39. Berard AM, Foger B, Remaley A, Shamburek R, Vaisman BL, Talley G, et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med 1997;3:744–749. ArticlePubMedPDF
- 40. Sakai N, Vaisman BL, Koch CA, Hoyt RF Jr, Meyn SM, Talley GD, et al. Targeted disruption of the mouse lecithin: cholesterol acyltransferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J Biol Chem 1997;272:7506–7510. ArticlePubMed
- 41. Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M, Rubin EM. Disruption of the murine lecithin:cholesterol acyltransferase gene causes impairment of adrenal lipid delivery and up-regulation of scavenger receptor class B type I. J Biol Chem 1997;272:15777–15781. ArticlePubMed
- 42. Hoeg JM, Vaisman BL, Demosky SJ Jr, Meyn SM, Talley GD, Hoyt RF Jr, et al. Lecithin:cholesterol acyltransferase overexpression generates hyperalpha-lipoproteinemia and a nonatherogenic lipoprotein pattern in transgenic rabbits. J Biol Chem 1996;271:4396–4402. ArticlePubMed
- 43. Hoeg JM, Santamarina-Fojo S, Berard AM, Cornhill JF, Herderick EE, Feldman SH, et al. Overexpression of lecithin: cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci U S A 1996;93:11448–11453. ArticlePubMedPMC
- 44. Holleboom AG, Kuivenhoven JA, Vergeer M, Hovingh GK, van Miert JN, Wareham NJ, et al. Plasma levels of lecithin:cholesterol acyltransferase and risk of future coronary artery disease in apparently healthy men and women: a prospective case-control analysis nested in the EPIC-Norfolk population study. J Lipid Res 2010;51:416–421. ArticlePubMedPMC
- 45. Calabresi L, Baldassarre D, Simonelli S, Gomaraschi M, Amato M, Castelnuovo S, et al. Plasma lecithin:cholesterol acyltransferase and carotid intima-media thickness in European individuals at high cardiovascular risk. J Lipid Res 2011;52:1569–1574. ArticlePubMedPMC
- 46. Dullaart RP, Perton F, Sluiter WJ, de Vries R, van Tol A. Plasma lecithin:cholesterol acyltransferase activity is elevated in metabolic syndrome and is an independent marker of increased carotid artery intima media thickness. J Clin Endocrinol Metab 2008;93:4860–4866. ArticlePubMedPDF
- 47. Dullaart RP, Perton F, van der Klauw MM, Hillege HL, Sluiter WJ. PREVEND Study Group. High plasma lecithin: cholesterol acyltransferase activity does not predict low incidence of cardiovascular events: possible attenuation of cardioprotection associated with high HDL cholesterol. Atherosclerosis 2010;208:537–542. ArticlePubMed
- 48. Tani S, Takahashi A, Nagao K, Hirayama A. Association of lecithin-cholesterol acyltransferase activity measured as a serum cholesterol esterification rate and low-density lipoprotein heterogeneity with cardiovascular risk: a cross-sectional study. Heart Vessels 2015 4 19 [Epub]. ArticlePDF
- 49. Hovig T, Gjone E. Familial plasma lecithin:cholesterol acyltransferase (LCAT) deficiency. Ultrastructural aspects of a new syndrome with particular reference to lesions in the kidneys and the spleen. Acta Pathol Microbiol Scand A 1973;81:681–697. PubMed
- 50. Wells IC, Peitzmeier G, Vincent JK. Lecithin:cholesterol acyltransferase and lysolecithin in coronary atherosclerosis. Exp Mol Pathol 1986;45:303–310. ArticlePubMed
- 51. Sethi AA, Sampson M, Warnick R, Muniz N, Vaisman B, Nordestgaard BG, et al. High pre-beta1 HDL concentrations and low lecithin:cholesterol acyltransferase activities are strong positive risk markers for ischemic heart disease and independent of HDL-cholesterol. Clin Chem 2010;56:1128–1137. ArticlePubMedPMCPDF
- 52. Dullaart RP, Tietge UJ, Kwakernaak AJ, Dikkeschei BD, Perton F, Tio RA. Alterations in plasma lecithin:cholesterol acyltransferase and myeloperoxidase in acute myocardial infarction: implications for cardiac outcome. Atherosclerosis 2014;234:185–192. ArticlePubMed
References
Fig. 1
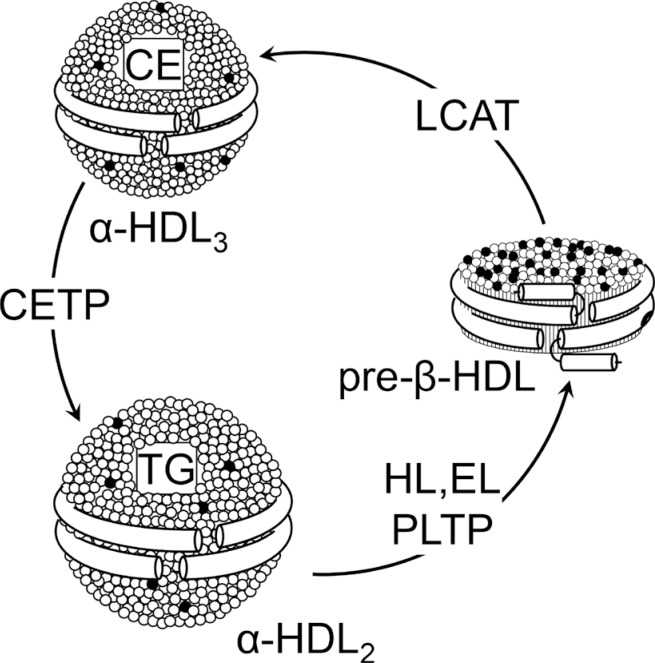
High-density lipoprotein (HDL) metabolism. HDL are a heterogeneous lipoprotein family and plasma interconversion within different subclasses is due to action of various enzymes. Lecithin:cholesterol acyltransferase (LCAT) esterifies free cholesterol located on the surface of pre-β-HDL. The cholesterol esters generated by LCAT activity are more hydrophobic than free cholesterol and they migrate into the hydrophobic core of the lipoprotein, with the resulting conversion of pre-β-HDL into α-HDL3. The α-HDL3 produced by LCAT interact with cholesteryl ester transfer protein (CETP), that exchanges cholesteryl esters for triglycerides generating large cholesteryl ester-poor and triglyceride-rich HDL particles (α-HDL2). α-HDL2 can be converted back to pre-β-HDL through a variety of lipases activities that hydrolyze triglycerides and phospholipids on HDL. CE, cholesteryl esters; TG, triglyceride; HL, hepatic lipase; EL, endothelial lipase; PLTP, phospholipid transfer protein.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Association between Mycoplasma pneumoniae infection, high‑density lipoprotein metabolism and cardiovascular health (Review)
Tao Shen, Yanfang Li, Tingting Liu, Yunzhi Lian, Luke Kong
Biomedical Reports.2024;[Epub] CrossRef - The Link between Magnesium Supplements and Statin Medication in Dyslipidemic Patients
Roxana Nartea, Brindusa Ilinca Mitoiu, Ioana Ghiorghiu
Current Issues in Molecular Biology.2023; 45(4): 3146. CrossRef - Mannose-Coated Reconstituted Lipoprotein Nanoparticles for the Targeting of Tumor-Associated Macrophages: Optimization, Characterization, and In Vitro Evaluation of Effectiveness
Akpedje S. Dossou, Morgan E. Mantsch, Ammar Kapic, William L. Burnett, Nirupama Sabnis, Jeffery L. Coffer, Rance E. Berg, Rafal Fudala, Andras G. Lacko
Pharmaceutics.2023; 15(6): 1685. CrossRef - Abdominal obesity negatively influences key metrics of reverse cholesterol transport
Jennifer Härdfeldt, Marica Cariello, Sara Simonelli, Alice Ossoli, Natasha Scialpi, Marilidia Piglionica, Emanuela Pasculli, Alessia Noia, Elsa Berardi, Patrizia Suppressa, Giuseppina Piazzolla, Carlo Sabbà, Laura Calabresi, Antonio Moschetta
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2022; 1867(2): 159087. CrossRef - Effects of Lactobacillus paracasei N1115 on dyslipidaemia: A randomized controlled study
Hua Jiang, Shengjie Tan, Ke Ning, Hao Li, Wenzhi Zhao, Ai Zhao, Hong Zhu, Shijie Wang, Peiyu Wang, Yumei Zhang
Journal of Functional Foods.2022; 89: 104956. CrossRef - LCAT- targeted therapies: Progress, failures and future
Kaixu Yang, Junmin Wang, Hongjiao Xiang, Peilun Ding, Tao Wu, Guang Ji
Biomedicine & Pharmacotherapy.2022; 147: 112677. CrossRef - Cyclodextrin boostered-high density lipoprotein for antiatherosclerosis by regulating cholesterol efflux and efferocytosis
Yanyan Wang, Hai Gao, Xinya Huang, Zhaoan Chen, Pengyu Kang, Yunyi Zhou, Danhua Qin, Wenli Zhang, Jianping Liu
Carbohydrate Polymers.2022; 292: 119632. CrossRef - Lipidomic Approaches to Study HDL Metabolism in Patients with Central Obesity Diagnosed with Metabolic Syndrome
Gabriele Mocciaro, Simona D’Amore, Benjamin Jenkins, Richard Kay, Antonio Murgia, Luis Vicente Herrera-Marcos, Stefanie Neun, Alice P. Sowton, Zoe Hall, Susana Alejandra Palma-Duran, Giuseppe Palasciano, Frank Reimann, Andrew Murray, Patrizia Suppressa, C
International Journal of Molecular Sciences.2022; 23(12): 6786. CrossRef - rHDL modeling and the anchoring mechanism of LCAT activation
Tommaso Laurenzi, Chiara Parravicini, Luca Palazzolo, Uliano Guerrini, Elisabetta Gianazza, Laura Calabresi, Ivano Eberini
Journal of Lipid Research.2021; 62: 100006. CrossRef - Vasculoprotective properties of plasma lipoproteins from brown bears (Ursus arctos)
Matteo Pedrelli, Paolo Parini, Jonas Kindberg, Jon M. Arnemo, Ingemar Bjorkhem, Ulrika Aasa, Maria Westerståhl, Anna Walentinsson, Chiara Pavanello, Marta Turri, Laura Calabresi, Katariina Öörni, Gérman Camejo, Ole Fröbert, Eva Hurt-Camejo
Journal of Lipid Research.2021; 62: 100065. CrossRef - The Fate of Dietary Cholesterol in the Kissing Bug Rhodnius prolixus
Petter F. Entringer, David Majerowicz, Katia C. Gondim
Frontiers in Physiology.2021;[Epub] CrossRef - Excesive consumption of unsaturated fatty acids leads to oxidative and inflammatory instability in Wistar rats
Jelica D. Grujić-Milanović, Zoran Z. Miloradović, Nevena D. Mihailović-Stanojević, Vojislav V. Banjac, Strahinja Vidosavljević, Milan S. Ivanov, Danijela J. Karanović, Una-Jovana V. Vajić, Djurdjica M. Jovović
Biomedicine & Pharmacotherapy.2021; 139: 111691. CrossRef - Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy
Isabella Giacomini, Federico Gianfanti, Maria Andrea Desbats, Genny Orso, Massimiliano Berretta, Tommaso Prayer-Galetti, Eugenio Ragazzi, Veronica Cocetta
Frontiers in Oncology.2021;[Epub] CrossRef - Association between Serum Concentrations of Apolipoprotein A-I (ApoA-I) and Alzheimer’s Disease: Systematic Review and Meta-Analysis
Marco Zuin, Carlo Cervellati, Alessandro Trentini, Angelina Passaro, Valentina Rosta, Francesca Zimetti, Giovanni Zuliani
Diagnostics.2021; 11(6): 984. CrossRef - Dapagliflozin reduces thrombin generation and platelet activation: implications for cardiovascular risk reduction in type 2 diabetes mellitus
Christina Kohlmorgen, Stephen Gerfer, Kathrin Feldmann, Sören Twarock, Sonja Hartwig, Stefan Lehr, Meike Klier, Irena Krüger, Carolin Helten, Petra Keul, Sabine Kahl, Amin Polzin, Margitta Elvers, Ulrich Flögel, Malte Kelm, Bodo Levkau, Michael Roden, Jen
Diabetologia.2021; 64(8): 1834. CrossRef - Impact of Dietary Lipids on the Reverse Cholesterol Transport: What We Learned from Animal Studies
Bianca Papotti, Joan Carles Escolà-Gil, Josep Julve, Francesco Potì, Ilaria Zanotti
Nutrients.2021; 13(8): 2643. CrossRef - Supramolecular copolymer modified statin-loaded discoidal rHDLs for atherosclerotic anti-inflammatory therapy by cholesterol efflux and M2 macrophage polarization
Qiqi Zhang, Jianhua He, Fengfei Xu, Xinya Huang, Yanyan Wang, Wenli Zhang, Jianping Liu
Biomaterials Science.2021; 9(18): 6153. CrossRef - Association between Small Dense Low-Density Lipoproteins and High-Density Phospolipid Content in Patients with Coronary Artery Disease with or without Diabetes
Hanene Aoua, Ymène Nkaies, Ali Ben Khalfallah, Mohsen Sakly, Ezzedine Aouani, Nebil Attia
Laboratory Medicine.2020; 51(3): 271. CrossRef - Biomechanical and biochemical investigation of erythrocytes in late stage human leptospirosis
J.A.X. Silva, A.V.P. Albertini, C.S.M. Fonseca, D.C.N. Silva, V.C.O. Carvalho, V.L.M. Lima, A. Fontes, E.V.L. Costa, R.A. Nogueira
Brazilian Journal of Medical and Biological Research.2020;[Epub] CrossRef - Cardioprotective Properties of HDL: Structural and Functional Considerations
Eleni Pappa, Moses S. Elisaf, Christina Kostara, Eleni Bairaktari, Vasilis K. Tsimihodimos
Current Medicinal Chemistry.2020; 27(18): 2964. CrossRef - Relationship between non–high-density lipoprotein cholesterol/apolipoprotein A-I and monocyte/high-density lipoprotein cholesterol ratio and coronary heart disease
Ya Li, Shu Li, Yulin Ma, Jialing Li, Mingying Lin, Jing Wan
Coronary Artery Disease.2020; 31(7): 623. CrossRef - Lipid transfer to high‐density lipoproteins in coronary artery disease patients with and without previous cerebrovascular ischemic events
Carlos J.D.G. Barbosa, Raul C. Maranhão, Renata S. Barreiros, Fatima R. Freitas, André Franci, Célia M.C. Strunz, Flávia B.B. Arantes, Thauany M. Tavoni, José A.F. Ramires, Roberto Kalil Filho, José C. Nicolau
Clinical Cardiology.2019; 42(11): 1100. CrossRef - Antibodies Against the C-Terminus of ApoA-1 Are Inversely Associated with Cholesterol Efflux Capacity and HDL Metabolism in Subjects with and without Type 2 Diabetes Mellitus
Robin Dullaart, Sabrina Pagano, Frank Perton, Nicolas Vuilleumier
International Journal of Molecular Sciences.2019; 20(3): 732. CrossRef - The effect of chronic kidney disease on lipid metabolism
Neris Dincer, Tuncay Dagel, Baris Afsar, Adrian Covic, Alberto Ortiz, Mehmet Kanbay
International Urology and Nephrology.2019; 51(2): 265. CrossRef - Biological Consequences of Dysfunctional HDL
Angela Pirillo, Alberico Luigi Catapano, Giuseppe Danilo Norata
Current Medicinal Chemistry.2019; 26(9): 1644. CrossRef - Plasma lecithin:cholesterol acyltransferase and phospholipid transfer protein activity independently associate with nonalcoholic fatty liver disease
Karlijn J. Nass, Eline H. van den Berg, Eke G. Gruppen, Robin P. F. Dullaart
European Journal of Clinical Investigation.2018;[Epub] CrossRef - An integrated metabolomic strategy for the characterization of the effects of Chinese yam and its three active components on septic cardiomyopathy
Ning Zhou, Meng-Nan Zeng, Kai Li, Yan-Yun Yang, Zhi-Yao Bai, Xiao-Ke Zheng, Wei-Sheng Feng
Food & Function.2018; 9(9): 4989. CrossRef - Effect of Rosuvastatin on Cholesterol Efflux Capacity and Endothelial Function in Type 2 Diabetes Mellitus and Dyslipidemia
Kyong Yeun Jung, Kyoung Min Kim, Sun Kyoung Han, Han Mi Yun, Tae Jung Oh, Sung Hee Choi, Kyong Soo Park, Hak Chul Jang, Soo Lim
Circulation Journal.2018; 82(5): 1387. CrossRef - Association Between Serum LDL-C and ApoB and SYNTAX Score in Patients With Stable Coronary Artery Disease
Taiwu Lin, Luzhao Wang, Jingbin Guo, Peng Liu, Liheng Chen, Mengqiu Wei, Gongxin Li
Angiology.2018; 69(8): 724. CrossRef - Influence of i.v. lipid emulsion on lipoprotein subclass in preterm infants
Hiroki Suganuma, Naho Ikeda, Natsuki Ohkawa, Hiromichi Shoji, Toshiaki Shimizu
Pediatrics International.2018; 60(9): 839. CrossRef - The HDL cholesterol/apolipoprotein A-I ratio: an indicator of cardiovascular disease
Eun-Jung Rhee, Christopher D. Byrne, Ki-Chul Sung
Current Opinion in Endocrinology, Diabetes & Obesity.2017; 24(2): 148. CrossRef - Moringa Leaves Prevent Hepatic Lipid Accumulation and Inflammation in Guinea Pigs by Reducing the Expression of Genes Involved in Lipid Metabolism
Manal Almatrafi, Marcela Vergara-Jimenez, Ana Murillo, Gregory Norris, Christopher Blesso, Maria Fernandez
International Journal of Molecular Sciences.2017; 18(7): 1330. CrossRef
 PubReader
PubReader Cite
Cite

