
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 31(1); 2016 > Article
-
Review ArticleSmall Heterodimer Partner and Innate Immune Regulation
-
Jae-Min Yuk1,2
 , Hyo Sun Jin2,3, Eun-Kyeong Jo2,3
, Hyo Sun Jin2,3, Eun-Kyeong Jo2,3 -
Endocrinology and Metabolism 2016;31(1):17-24.
DOI: https://doi.org/10.3803/EnM.2016.31.1.17
Published online: March 16, 2016
1Department of Infection Biology, Chungnam National University School of Medicine, Daejeon, Korea.
2Infection Signaling Network Research Center, Chungnam National University School of Medicine, Daejeon, Korea.
3Department of Microbiology, Chungnam National University School of Medicine, Daejeon, Korea.
- Corresponding author: Eun-Kyeong Jo. Department of Microbiology, and Infection Signaling Network Research Center, Chungnam National University School of Medicine, 266 Munhwa-ro, Jung-gu, Daejeon 35015, Korea. Tel: +82-42-580-8243, Fax: +82-42-585-3686, hayoungj@cnu.ac.kr
• Received: December 14, 2015 • Revised: December 21, 2015 • Accepted: December 31, 2015
Copyright © 2016 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The nuclear receptor superfamily consists of the steroid and non-steroid hormone receptors and the orphan nuclear receptors. Small heterodimer partner (SHP) is an orphan family nuclear receptor that plays an essential role in the regulation of glucose and cholesterol metabolism. Recent studies reported a previously unidentified role for SHP in the regulation of innate immunity and inflammation. The innate immune system has a critical function in the initial response against a variety of microbial and danger signals. Activation of the innate immune response results in the induction of inflammatory cytokines and chemokines to promote anti-microbial effects. An excessive or uncontrolled inflammatory response is potentially harmful to the host, and can cause tissue damage or pathological threat. Therefore, the innate immune response should be tightly regulated to enhance host defense while preventing unwanted immune pathologic responses. In this review, we discuss recent studies showing that SHP is involved in the negative regulation of toll-like receptor-induced and NLRP3 (NACHT, LRR and PYD domains-containing protein 3)-mediated inflammatory responses in innate immune cells. Understanding the function of SHP in innate immune cells will allow us to prevent or modulate acute and chronic inflammation processes in cases where dysregulated innate immune activation results in damage to normal tissues.
- Nuclear receptors (NRs) are transcriptional regulators of a diverse set of genes that play a central role in metabolic and endocrine homeostasis. NRs play key physiological roles in the regulation of gene networks that operate a variety of biological responses, including cellular growth and differentiation, development, cell cycle and proliferation, and metabolism [123]. Consequently, NRs and their modulating agents are emerging as promising therapeutic targets in various metabolic, reproductive, and proliferative disorders [3]. Most NRs are activated by endogenous ligands, but others have been classified as orphan NRs without known ligands [4]. Small heterodimer partner (SHP, NR0B2) is an atypical orphan NR, since the endogenous ligand has not been identified, with a unique structure and function that are distinct from conventional NRs [56].
- In response to pathogenic or dangerous stimulation, the innate immune system is activated by triggering its numerous pattern-recognition receptors (PRRs) on innate cells, and induces pro-inflammatory and antimicrobial responses via intracellular signaling cascades [789]. It is clear that this PRR-mediated innate immune signaling is essential for the orchestration of the immediate host defense to infection and the subsequent activation of adaptive immunity [1011]. To avoid harmful immunopathological responses, the innate immune system is tightly regulated by a variety of molecules/pathways that control the magnitude of the inflammatory responses [1011].
- Recently, we showed that SHP is an essential negative regulator of innate immune signaling and suppresses toll-like receptor (TLR) and NLRP3 (NACHT [NOD, NAIP, CIITA, HET-E, or TP1], leucine-rich repeat [LRR], and PYD domains-containing protein 3)-induced inflammasome activation in macrophages and in mice [121314]. The aim of this brief review was to discuss the unexpected role of SHP in macrophages in light of recent data on the molecular mechanisms by which SHP controls TLR-induced inflammation and NLRP3 inflammasome activation.
INTRODUCTION
- The SHP gene consists of two exons and a single intron located at human chromosome 1p36.1 subband [15]. The human SHP gene is expressed in major organs and tissues, including the liver, heart, pancreas, kidney, adrenal gland, spleen, stomach, and small intestine [151617]. The structure of SHP is relatively unique because it lacks a conventional DNA-binding domain, but has a putative ligand-binding domain, which makes SHP a member of the NR family [518]. Previous studies have shown that SHP exerts its transcriptional regulatory function through protein-protein interactions with a variety of NRs [61920]. For example, SHP interacts with several NR family members, including retinoid receptors, the thyroid hormone receptor, and the orphan receptor MB67 [5], etc. Through these interactions, SHP can affect diverse biological responses, including cholesterol, bile acid, triglyceride, and glucose homeostasis [62021].
- Although SHP has no physiological ligands, numerous NRs and transcription factors (TFs) are able to target the SHP promoter and induce the expression of SHP. Earlier reports showed that steroidogenic factor 1 and the orphan fetoprotein transcription factor/liver receptor homologue 1 (LRH-1) can potently upregulate SHP promoter activity in tissues such as the adrenal glands and liver [22]. The farsenoid X receptor (FXR) was also shown to induce SHP transcription, which then inactivates LRH-1 to repress the expression of CYP7A1, the rate-limiting enzyme in the bile acid biosynthesis pathway; thus, resulting in the coordinated regulation of hepatic cholesterol metabolism [2324]. In addition, hepatocyte nuclear factor 4α (HNF4α) was also reported to regulate the transcription of FXR to inhibit the expression of SHP, which also heterodimerizes with HNF4α and inhibits its transcriptional activation [2526]. Moreover, other TFs, including c-Jun, basic helix-loop-helix TFs, and in particular the E2A proteins (E47, E12, and E2/5), function to activate SHP promoter activity [2728]. In addition, estrogen receptor-related receptor γ [16], liver X receptor α (LXRα) [29], peroxisome proliferator-activated receptor γ (PPAR-γ) [30], hepatocyte growth factor, and its family member macrophage-stimulating factor (MSP) were also involved in modulation of SHP promoter activity through upstream stimulatory factor 1 [31].
- Several genetic variations, including single nucleotide polymorphisms and mutations in the human SHP gene, have been reported in relation to a variety of physiological responses [20]. A previous report showed SHP mutations in Japanese patients with obesity and diabetes, suggesting roles for SHP in insulin resistance and mild obesity [32]. In addition, SHP mutations identified in mildly obese subjects were found to be associated with an increased risk for type 2 diabetes later in life in Japanese patients [33]. However, the relationship between SHP genetic variations with diabetes or obesity has been controversial in European studies. Novel SHP missense mutations and polymorphisms were identified in subjects with severe obesity in the UK [34]. However, another study with 1,927 UK subjects showed no specific relationship between SHP genetic variation and type 2 diabetes, obesity, or birth weight [35]. In a cohort study of Danish people with early-onset obesity, several novel SHP variants were identified [36]. Therefore, these findings suggest the need for large-scale population studies to assess the relevant risk of human diseases in relation to SHP gene mutations and/or polymorphisms.
OVERVIEW OF SHP STRUCTURE AND FUNCTION
- TLRs and nucleotide binding oligomerization domain (NOD)-like receptors (NLRs) are well-characterized membrane-bound and cytosolic PRRs, respectively [937]. They play important roles in innate recognition and inflammatory response elicitation against invading pathogens [9]. Both receptors have LRRs that are responsible for ligand interactions.
- TLRs are transmembrane innate PRRs located at the cell surface and in the intracellular membrane. So far, 12 mouse and 10 human functional TLR members have been identified [38]. Each TLR can sense distinct pathogen-associated molecular patterns (PAMPs) from various pathogens, including lipoproteins (by TLR2/1 and TLR2/6), double-stranded RNA (by TLR3), and lipopolysaccharide (LPS; by TLR4) [38]. Other TLRs, including TLR3, 7, 8, and 9, are present on intracellular membranes and can recognize bacterial and viral nucleic acids [38]. TLR engagement by PAMP or damage-associated molecular patterns (DAMP) initiates overlapping and distinct signaling pathways in innate immune cells [938]. There are several principal adaptor proteins, including MyD88, MyD88 adapter-like (MAL), Toll/IL-1 receptor domain-containing adaptor inducing β (TRIF), and Trif-related adaptor molecule (TRAM), that contain Toll/interleukin-1 receptor homology (TIR) domains and mediate the intracellular signaling triggered by TLRs [3940]. Most TLRs, except for TLR3, are able to transmit signals through the adaptor protein MyD88, whereas TLR3 and TLR4 signal through TRIF [383940].
- Twenty-two NLR family members that contain a C-terminal LRR domain, a central NACHT domain, and an N-terminal effector motif have been described [41]. NOD1 and NOD2 detect diaminopimelatic acid-containing muropeptide, primarily found in gram-negative bacteria, and muramyl dipeptide (MDP) moieties, usually found in all bacterial peptidoglycans, respectively, to initiate an innate inflammatory response and host defense pathways [4243444546]. Pyrin domain-containing proteins (NLRPs) are another group of NLRs; of these NLRs, NLRP3 is the best characterized, and activates inflammasome assembly [46].
- Inflammasomes are an intracellular molecular platform of multiprotein complexes that activate caspase-1, resulting in the maturation of pro-interleukin-1β (pro-IL-1β) to IL-1β and its subsequent release [47484950]. Numerous PAMP and DAMP signals can activate the NLRP3 inflammasome complex, including adenosine triphosphate, MDP, uric acid crystals, cholesterol crystals, silica, and specific pathogenic infections [4649505152]. Indeed, NLRP3 inflammasome activation is mediated through common general molecular pathways such as increased potassium efflux, lysosomal damage, or mitochondrial reactive oxygen species (ROS) generation [46535455]. Currently, a two-step process is generally accepted for NLRP3 inflammasome activation. The first "priming" step involves nuclear factor κB (NF-κB)-dependent production of inactive precursors pro-IL-1β and pro-IL-18 and inflammasome components, including NLRP3 [56]. Following activation, NLRP3 can assemble the caspase-1-dependent inflammasome by recruiting an adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), also known as TMS1 or PYCARD, and procaspase-1 [465354]. A second signal is required for NLRP3 inflammasome assembly by the recruitment of ASC and procaspase-1 to activate caspase-1 and to release mature cytokines IL-1β and IL-18 [465354].
- Previous reports have shown that NLRP3 mutations are involved in an autoinflammatory disease, cryopyrin-associated periodic syndrome, which manifests as systemic inflammation and serious complications [57]. Therapeutic intervention using IL-1β neutralization is highly beneficial in these patients [57]. Accumulating evidence also suggests that NLRP3 is involved in inflammatory bowel disease, cardiovascular disease, and rheumatoid arthritis [58]. In addition, numerous studies showed that NLRP3 inflammasome activation is important for the pathogenesis of metabolic syndromes, such as insulin resistance and obesity [59]. Moreover, dysregulated activation of the NLRP3 inflammasome seems to be implicated in the pathogenesis of other chronic inflammatory diseases, including Alzheimer disease [60], atherosclerosis [61], and age-related macular degeneration [6263]. Therefore, recent efforts to modulate NLRP3 inflammasome complex activation may create new opportunities to treat or prevent numerous chronic inflammatory and metabolic diseases.
INNATE IMMUNITY: TLRs AND THE INFLAMMASOME
- Since innate immune activation should be tightly regulated to prevent unwanted host damage, numerous studies have identified the roles of negative regulators in TLR signaling and NLRP3 inflammasome activation [6465]. Recently, we described a previously unknown role for SHP in the negative regulation of TLR signaling [1314]. During TLR engagement, proinflammatory cytokines are produced by a series of intracellular mediators including interleukin-1 receptor-associated kinase 1, TNF receptor-associated factor 6 (TRAF6), and NF-κB in macrophages. SHP is essential for the regulation of TLR-mediated transactivation of canonical NF-κB and Lys63-linked polyubiquitination of TRAF6. In unstimulated cells, SHP is associated with the NF-κB p65 subunit in the cytoplasm and inhibits the nuclear translocation of NF-κB p65, resulting in the attenuation of proinflammatory transcriptional activity. TLR4 stimulation leads to rapid dissociation of SHP from the NF-κB p65 subunit, and SHP then interacts with the TRAF6 really interesting new gene (RING) domain that plays a crucial role in TRAF6 ubiquitination and activation [13].
- Our previous studies demonstrated the possibility of SHP-inducing drugs as therapeutic candidates for systemic inflammatory diseases. Although SHP-specific ligands have not yet been identified, treatment with various AMP-activated protein kinase (AMPK)-activating drugs, such as MSP and fenofibrates, induce the expression of SHP in hepatocytes [3166] and macrophages [1314]. In addition, we demonstrated that liver kinase B1-dependent AMPK activation is required for the expression of SHP in macrophages treated with MSP and fenofibrate. Several studies have shown that MSP plays an inhibitory role in the LPS-mediated macrophage production of nitric oxide [6768], and the mechanisms by which MSP proteins are involved in TLR signaling involve the attenuation of serine phosphorylation of NF-κB p65 and NF-κB-dependent activation of IκBζ [69]. Our study further showed that MSP has a regulatory function in TLR4-mediated inflammatory responses through prevention of TRAF6 polyubiquitination. Moreover, fenofibrate is a well-known ligand of PPARα that has an anti-inflammatory effect in acute respiratory distress syndrome [70] and retinal hyperinflammation of type 1 diabetes-induced retinopathy [71]. However, fenofibrate improves survival in mice and inhibits the production of proinflammatory cytokines and mitochondrial ROS through PPARα-independent AMPK-SHP-uncoupling protein 2 (UCP2) regulatory cascades in TLR4-mediated systemic inflammatory responses. These data strongly suggest that the development and discovery of potential candidates involved in SHP induction and activity modulation may partially control systemic inflammatory diseases.
- Previous studies suggested that the inflammasome plays a crucial role in host immune defense during infection with a variety of pathogens such as microbes, virus, and parasites [727374]. However, uncontrolled activation of inflammasomes leads to autoinflammatory diseases that are associated with the production of inflammatory cytokines such as IL-1β [7576]. We recently identified an unrecognized role for SHP in the regulation of the NLRP3 inflammasome [12]. SHP deficiency results in enhanced activation of the NLRP3 inflammasome, while abundant SHP protein produced by SHP-inducing agents, including MSP and fenofibrate, competitively inhibits the physical interaction between NLRP3 and ASC, and then intercepts the assembly of the NLRP3 inflammasome complex, which eventually leads to down-regulation of IL-1β and IL-18 production in NLRP3-activated macrophages. Moreover, SHP is translocated to the mitochondria with the NLRP3-ASC complex after inflammasome activation and then acts to regulate mitochondrial homeostasis through the recovery of inflammasome-induced mitochondrial ROS generation and damage. These findings demonstrate that SHP plays a fine-tuning role to prevent an excessive inflammatory response (Fig. 1).
SHP REGULATION OF INNATE IMMUNITY AND INFLAMMATION
- Accumulating evidence strongly suggests that NRs are crucial regulators of the physiological responses related to the progression of various diseases, including inflammatory and metabolic diseases. Because an uncontrolled or excessive inflammatory response generally leads to venomous pathogenesis with a high mortality rate, many lines of research have focused on the negative regulation of this process, and numerous negative regulators have been identified as a result. Several NRs, such as the glucocorticoid receptor, pregnane X receptor, PPAR-γ, and LXRs, are involved in the regulation of TLR signaling through post-translational or transcriptional repression. In this issue, we focus on the regulatory roles of orphan NR SHP during activation of the TLR-mediated inflammatory response and NLRP3 inflammasome activity. We previously demonstrated that the regulation of TLR signaling and NLRP3 inflammasome activity by SHP and gene expression of SHP are closely related, which suggests functional autoregulation of SHP during the innate immune response. In brief, SHP interacts with cytosolic p65 in resting cells, whereas TLR stimulation leads to rapid dissociation between SHP and p65. The cytosolic SHP protein then interacts with TRAF6, thereby completing the polyubiquitination of TRAF6. Moreover, SHP deficiency results in enhanced generation of mitochondrial ROS that is related to the decrease in mitochondrial anion carrier protein UCP2 protein expression. During activation of the NLRP3 inflammasome, SHP temporarily interacts with endogenous NLRP3 in the perinuclear region, translocates to the mitochondria to maintain mitochondrial homeostasis, and then attenuates caspase-1 activation and IL-1β and IL-18 cytokine maturation. Taken together, these studies indicate that SHP is an intrinsic negative regulator of NLRP3 inflammasome and TLR-mediated inflammatory responses. Moreover, our results strongly suggest that therapeutic or protective drugs, such as MSP and fenofibrate, which modulate SHP induction and activity, partially control inflammatory diseases including endotoxic shock and acute tubular necrosis. Understanding the roles of SHP and various NRs in animals and humans will facilitate the development of novel therapeutics to treat inflammatory diseases.
CONCLUSIONS
-
Acknowledgements
- We are indebted to current and past members of our laboratory for discussions and investigations that contributed to this article. This work was supported by research fund of Chungnam National University and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP; No. NRF-2015M3C9A2054326). I apologize to colleagues whose work and publications could not be referenced owing to space constraints.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, et al. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 2006;58:760–772. ArticlePubMed
- 2. Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 2004;3:950–964. ArticlePubMedPDF
- 3. Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K, et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev 2013;65:710–778. ArticlePubMed
- 4. Ranhotra HS. The orphan estrogen-related receptor alpha and metabolic regulation: new frontiers. J Recept Signal Transduct Res 2015;35:565–568. ArticlePubMed
- 5. Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science 1996;272:1336–1339. ArticlePubMed
- 6. Lee YS, Chanda D, Sim J, Park YY, Choi HS. Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int Rev Cytol 2007;261:117–158. ArticlePubMed
- 7. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 2009;22:240–273. ArticlePubMedPMC
- 8. Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell 1997;91:295–298. ArticlePubMed
- 9. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011;30:16–34. ArticlePubMed
- 10. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005;17:1–14. ArticlePubMedPDF
- 11. Coll RC, O'Neill LA. New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J Innate Immun 2010;2:406–421. ArticlePubMed
- 12. Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun 2015;6:6115ArticlePubMedPMCPDF
- 13. Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol 2011;12:742–751. ArticlePubMedPDF
- 14. Yang CS, Yuk JM, Kim JJ, Hwang JH, Lee CH, Kim JM, et al. Small heterodimer partner-targeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2. PLoS One 2013;8:e63435ArticlePubMedPMC
- 15. Lee HK, Lee YK, Park SH, Kim YS, Park SH, Lee JW, et al. Structure and expression of the orphan nuclear receptor SHP gene. J Biol Chem 1998;273:14398–14402. ArticlePubMed
- 16. Sanyal S, Kim JY, Kim HJ, Takeda J, Lee YK, Moore DD, et al. Differential regulation of the orphan nuclear receptor small heterodimer partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms. J Biol Chem 2002;277:1739–1748. ArticlePubMed
- 17. Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem 2002;277:1586–1592. ArticlePubMed
- 18. Seol W, Chung M, Moore DD. Novel receptor interaction and repression domains in the orphan receptor SHP. Mol Cell Biol 1997;17:7126–7131. ArticlePubMedPMC
- 19. Chanda D, Park JH, Choi HS. Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocr J 2008;55:253–268. ArticlePubMed
- 20. Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta 2011;1812:893–908. ArticlePubMed
- 21. Zhang Y, Wang L. Nuclear receptor small heterodimer partner in apoptosis signaling and liver cancer. Cancers (Basel) 2011;3:198–212. ArticlePubMedPMC
- 22. Lee YK, Parker KL, Choi HS, Moore DD. Activation of the promoter of the orphan receptor SHP by orphan receptors that bind DNA as monomers. J Biol Chem 1999;274:20869–20873. ArticlePubMed
- 23. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 2000;6:507–515. ArticlePubMed
- 24. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 2000;6:517–526. ArticlePubMed
- 25. Shih DQ, Screenan S, Munoz KN, Philipson L, Pontoglio M, Yaniv M, et al. Loss of HNF-1alpha function in mice leads to abnormal expression of genes involved in pancreatic islet development and metabolism. Diabetes 2001;50:2472–2480. ArticlePubMed
- 26. Shih DQ, Bussen M, Sehayek E, Ananthanarayanan M, Shneider BL, Suchy FJ, et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet 2001;27:375–382. ArticlePubMedPDF
- 27. Gupta S, Stravitz RT, Dent P, Hylemon PB. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J Biol Chem 2001;276:15816–15822. ArticlePubMed
- 28. Kim HJ, Kim JY, Park YY, Choi HS. Synergistic activation of the human orphan nuclear receptor SHP gene promoter by basic helix-loop-helix protein E2A and orphan nuclear receptor SF-1. Nucleic Acids Res 2003;31:6860–6872. ArticlePubMedPMC
- 29. Goodwin B, Watson MA, Kim H, Miao J, Kemper JK, Kliewer SA. Differential regulation of rat and human CYP7A1 by the nuclear oxysterol receptor liver X receptor-alpha. Mol Endocrinol 2003;17:386–394. ArticlePubMed
- 30. Kim HI, Koh YK, Kim TH, Kwon SK, Im SS, Choi HS, et al. Transcriptional activation of SHP by PPAR-gamma in liver. Biochem Biophys Res Commun 2007;360:301–306. ArticlePubMed
- 31. Chanda D, Li T, Song KH, Kim YH, Sim J, Lee CH, et al. Hepatocyte growth factor family negatively regulates hepatic gluconeogenesis via induction of orphan nuclear receptor small heterodimer partner in primary hepatocytes. J Biol Chem 2009;284:28510–28521. ArticlePubMedPMC
- 32. Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, Sho K, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci U S A 2001;98:575–580. ArticlePubMedPMC
- 33. Enya M, Horikawa Y, Kuroda E, Yonemaru K, Tonooka N, Tomura H, et al. Mutations in the small heterodimer partner gene increase morbidity risk in Japanese type 2 diabetes patients. Hum Mutat 2008;29:E271–E277. ArticlePubMed
- 34. Hung CC, Farooqi IS, Ong K, Luan J, Keogh JM, Pembrey M, et al. Contribution of variants in the small heterodimer partner gene to birthweight, adiposity, and insulin levels: mutational analysis and association studies in multiple populations. Diabetes 2003;52:1288–1291. ArticlePubMed
- 35. Mitchell SM, Weedon MN, Owen KR, Shields B, Wilkins-Wall B, Walker M, et al. Genetic variation in the small heterodimer partner gene and young-onset type 2 diabetes, obesity, and birth weight in U.K. subjects. Diabetes 2003;52:1276–1279. ArticlePubMed
- 36. Echwald SM, Andersen KL, Sorensen TI, Larsen LH, Andersen T, Tonooka N, et al. Mutation analysis of NR0B2 among 1545 Danish men identifies a novel c.278G>A (p.G93D) variant with reduced functional activity. Hum Mutat 2004;24:381–387. ArticlePubMed
- 37. Lee MS, Kim YJ. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol Cells 2007;23:1–10. ArticlePubMed
- 38. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011;34:637–650. ArticlePubMed
- 39. Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets 2012;13:1360–1374. ArticlePubMed
- 40. Watters TM, Kenny EF, O'Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol 2007;85:411–419. ArticlePubMed
- 41. Franchi L, Park JH, Shaw MH, Marina-Garcia N, Chen G, Kim YG, et al. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol 2008;10:1–8. ArticlePubMed
- 42. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 2003;4:702–707. ArticlePubMedPDF
- 43. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003;300:1584–1587. ArticlePubMed
- 44. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003;278:8869–8872. ArticlePubMed
- 45. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem 2003;278:5509–5512. ArticlePubMed
- 46. Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc Natl Acad Sci U S A 2014;111:12294–12299. ArticlePubMedPMC
- 47. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev 2011;243:136–151. ArticlePubMed
- 48. Lamkanfi M, Dixit VM. The inflammasomes. PLoS Pathog 2009;5:e1000510ArticlePubMedPMC
- 49. Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 2010;22:28–33. ArticlePubMedPMC
- 50. Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell 2006;126:659–662. ArticlePubMed
- 51. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013;13:397–411. ArticlePubMedPDF
- 52. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009;27:229–265. ArticlePubMed
- 53. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417–426. ArticlePubMed
- 54. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol 2011;166:1–15. ArticlePubMedPMC
- 55. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–225. ArticlePubMedPDF
- 56. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–791. ArticlePubMedPMC
- 57. Kone-Paut I, Galeotti C. Current treatment recommendations and considerations for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol 2015;11:1083–1092. ArticlePubMed
- 58. Paramel GV, Sirsjo A, Fransen K. Role of genetic alterations in the NLRP3 and CARD8 genes in health and disease. Mediators Inflamm 2015;2015:846782ArticlePubMedPMCPDF
- 59. Haneklaus M, O'Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev 2015;265:53–62. ArticlePubMed
- 60. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674–678. ArticlePubMedPDF
- 61. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–1361. ArticlePubMedPMCPDF
- 62. Celkova L, Doyle SL, Campbell M. NLRP3 Inflammasome and Pathobiology in AMD. J Clin Med 2015;4:172–192. ArticlePubMedPMC
- 63. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 2012;18:791–798. ArticlePubMedPMCPDF
- 64. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol 2012;33:449–458. ArticlePubMed
- 65. Chen S, Sun B. Negative regulation of NLRP3 inflammasome signaling. Protein Cell 2013;4:251–258. ArticlePubMedPMCPDF
- 66. Chanda D, Lee CH, Kim YH, Noh JR, Kim DK, Park JH, et al. Fenofibrate differentially regulates plasminogen activator inhibitor-1 gene expression via adenosine monophosphate-activated protein kinase-dependent induction of orphan nuclear receptor small heterodimer partner. Hepatology 2009;50:880–892. ArticlePubMedPMC
- 67. Liu QP, Fruit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN-gamma and lipopolysaccharide by the STK/RON receptor tyrosine kinase. J Immunol 1999;163:6606–6613. PubMed
- 68. Chen YQ, Fisher JH, Wang MH. Activation of the RON receptor tyrosine kinase inhibits inducible nitric oxide synthase (iNOS) expression by murine peritoneal exudate macrophages: phosphatidylinositol-3 kinase is required for RON-mediated inhibition of iNOS expression. J Immunol 1998;161:4950–4959. ArticlePubMedPDF
- 69. Ray M, Yu S, Sharda DR, Wilson CB, Liu Q, Kaushal N, et al. Inhibition of TLR4-induced IκB kinase activity by the RON receptor tyrosine kinase and its ligand, macrophage-stimulating protein. J Immunol 2010;185:7309–7316. ArticlePubMedPMC
- 70. Hecker M, Behnk A, Morty RE, Sommer N, Vadasz I, Herold S, et al. PPAR-α activation reduced LPS-induced inflammation in alveolar epithelial cells. Exp Lung Res 2015;41:393–403. ArticlePubMed
- 71. Chen Y, Hu Y, Lin M, Jenkins AJ, Keech AC, Mott R, et al. Therapeutic effects of PPARα agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013;62:261–272. ArticlePubMed
- 72. Yuk JM, Jo EK. Crosstalk between autophagy and inflammasomes. Mol Cells 2013;36:393–399. ArticlePubMedPMCPDF
- 73. Clay GM, Sutterwala FS, Wilson ME. NLR proteins and parasitic disease. Immunol Res 2014;59:142–152. ArticlePubMedPMCPDF
- 74. Kim JJ, Jo EK. NLRP3 inflammasome and host protection against bacterial infection. J Korean Med Sci 2013;28:1415–1423. ArticlePubMedPMC
- 75. Dinarello CA. Mutations in cryopyrin: bypassing roadblocks in the caspase 1 inflammasome for interleukin-1beta secretion and disease activity. Arthritis Rheum 2007;56:2817–2822. ArticlePubMed
- 76. Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum 2007;56:3138–3148. ArticlePubMed
References
Fig. 1
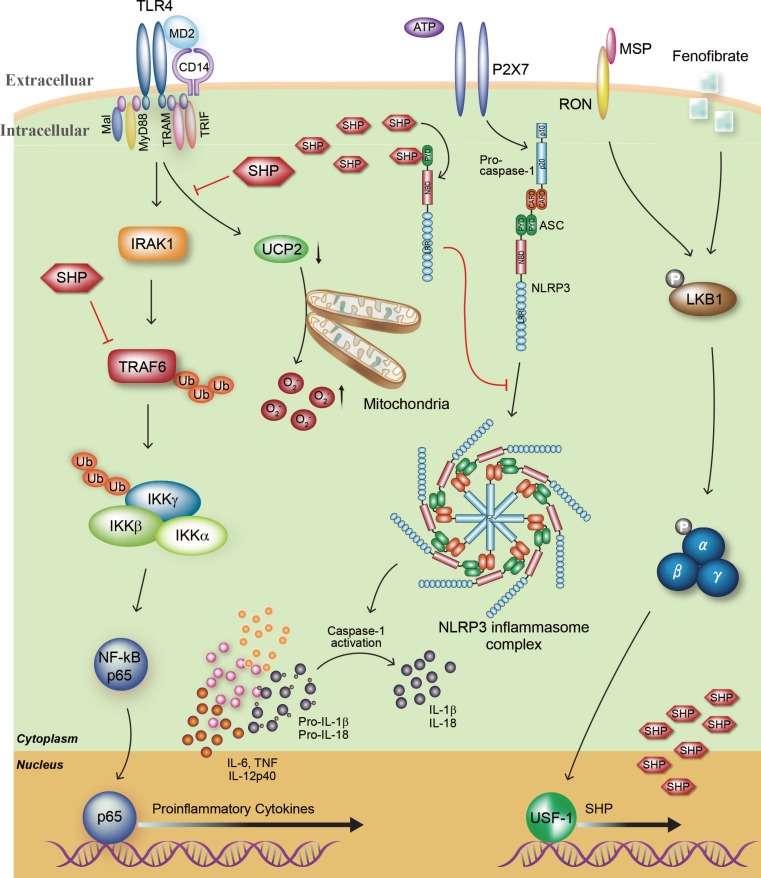
Roles of the orphan nuclear receptor small heterodimer partner (SHP) in the toll-like receptor 4 (TLR4)-mediated inflammatory response and NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome activity. The orphan nuclear receptor SHP plays an important role in the negative regulation of lipopolysaccharide (LPS)-meditated inflammation and NLRP3 inflammasome activation. First, TLR4 engagement strongly acts to generate proinflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), IL-1β, and IL-12p40 through TNF receptor-associated factor 6 (TRAF6) ubiquitination and nuclear factor κB (NF-κB) p65 nuclear translocation. Moreover, the generation of mitochondrial reactive oxygen species (ROS) in macrophages is enhanced during LPS stimulation, which is closely associated with activation of the inflammatory response. The mitochondrial anion carrier protein uncoupling protein 2 (UCP2) is an essential component of mitochondrial ROS and inflammation modulation. Our previous studies indicated that endogenous SHP inhibits the TLR4-induced upregulation of proinflammatory responses through the modulation of TRAF6 polyubiquitination and UCP2 expression. Second, inflammasome activation results in the maturation of pro-IL-1β and pro-IL-18 and the secretion of these mature cytokines into the extracellular environment. SHP deficiency causes enhanced secretion of mature IL-1β and IL-18. On the other hand, overexpression of SHP effectively attenuates NLRP3 activation and secretion of IL-1β and IL-18 through direct interaction with NLRP3 protein. Third, treatment with fenofibrate or macrophage-stimulating factor (MSP) activates SHP gene expression through liver kinase B1 (LKB1)-dependent AMP-activated protein kinase activation and recruitment of upstream stimulatory factor-1 (USF1) to the human SHP promoter. In addition, in vivo administration of SHP-inducing drugs effectively protects against LPS-induced lethal shock and folic acid-induced acute tubular necrosis. MD2, lymphocyte antigen 96; TRAM, Trif-related adaptor molecule; TRIF, Toll/IL-1 receptor domain-containing adaptor inducing β; RON, recepteur d'origine nantais; NBD, nucleotide-binding domain; CARD, caspase activation and recruitment domains; PYD, pyrin domain; ASC, adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain; IRAK1, interleukin-1 receptor-associated kinase 1; IKK, IκB kinase.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Upregulation of Anti-Angiogenic miR-106b-3p Correlates Negatively with IGF-1 and Vascular Health Parameters in a Model of Subclinical Cardiovascular Disease: Study with Metformin Therapy
Sherin Bakhashab, Josie O’Neill, Rosie Barber, Catherine Arden, Jolanta U. Weaver
Biomedicines.2024; 12(1): 171. CrossRef - Role of sodium taurocholate cotransporting polypeptide (NTCP) in HBV-induced hepatitis: Opportunities for developing novel therapeutics
Zhentao Zhang, Qi Zhang, Yiwen Zhang, Yutao Lou, Luqi Ge, Wanli Zhang, Wen Zhang, Feifeng Song, Ping Huang
Biochemical Pharmacology.2024; 219: 115956. CrossRef - Chlorpyrifos induces apoptosis and necroptosis via the activation of CYP450s pathway mediated by nuclear receptors in LMH cells
Xinyu Zhang, Kexin Sun, Xu Wang, Xu Shi, Duqiang Gong
Environmental Science and Pollution Research.2023; 30(1): 1060. CrossRef - A perspective study of the possible impact of obeticholic acid against SARS-CoV-2 infection
Gaber El-Saber Batiha, Hayder M. Al-kuraishy, Ali I. Al-Gareeb, Fadia S. Youssef, Suzy A. El-Sherbeni, Walaa A. Negm
Inflammopharmacology.2023; 31(1): 9. CrossRef - Is Nuclear Factor Erythroid 2-Related Factor 2 a Target for the Intervention of Cytokine Storms?
Zihang Liu, Panpan Deng, Shengnan Liu, Yiying Bian, Yuanyuan Xu, Qiang Zhang, Huihui Wang, Jingbo Pi
Antioxidants.2023; 12(1): 172. CrossRef - Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways
Forkan Ahamed, Natalie Eppler, Elizabeth Jones, Lily He, Yuxia Zhang
Biomedicines.2023; 11(9): 2403. CrossRef - Natural products modulate NLRP3 in ulcerative colitis
Jia-Chen Xue, Shuo Yuan, Xiao-Ting Hou, Huan Meng, Bao-Hong Liu, Wen-Wen Cheng, Ming Zhao, Hong-Ben Li, Xue-Fen Guo, Chang Di, Min-Jie Li, Qing-Gao Zhang
Frontiers in Pharmacology.2023;[Epub] CrossRef - The effectiveness of small heterodimer partner and FGF 19 levels in prediction of perinatal morbidity in intrahepatic cholestasis of pregnancy
Mehmet Bayram, Kader Irak, Sami Cifci, Ali Riza Koksal, Cemal Kazezoglu, Zuat Acar, Halil Onur Ozarı, Huseyin Alkim
Journal of Obstetrics and Gynaecology.2022; 42(5): 1174. CrossRef - Nuclear Receptors as Multiple Regulators of NLRP3 Inflammasome Function
Ahmad Alatshan, Szilvia Benkő
Frontiers in Immunology.2021;[Epub] CrossRef - The Orphan Nuclear Receptor Gene NR0B2 Is a Favorite Prognosis Factor Modulated by Multiple Cellular Signal Pathways in Human Liver Cancers
Runzhi Zhu, Yanjie Tu, Jingxia Chang, Haixia Xu, Jean C. Li, Wang Liu, Ahn-Dao Do, Yuxia Zhang, Jinhu Wang, Benyi Li
Frontiers in Oncology.2021;[Epub] CrossRef - Small heterodimer partner as a predictor of neoadjuvant radiochemotherapy response and survival in patients with rectal cancer: A preliminary study
Sup Kim, Mina Joo, Min-Kyung Yeo, Moon-June Cho, Jun-Sang Kim, Eun-Kyeong Jo, Jin-Man Kim
Oncology Letters.2021;[Epub] CrossRef - LPS Stimulation Induces Small Heterodimer Partner Expression Through the AMPK-NRF2 Pathway in Large Yellow Croaker (Larimichthys crocea)
Jianlong Du, Xiaojun Xiang, Dan Xu, Kun Cui, Yuning Pang, Wei Xu, Kangsen Mai, Qinghui Ai
Frontiers in Immunology.2021;[Epub] CrossRef - Host Transcription Factors in Hepatitis B Virus RNA Synthesis
Kristi L. Turton, Vanessa Meier-Stephenson, Maulik D. Badmalia, Carla S. Coffin, Trushar R. Patel
Viruses.2020; 12(2): 160. CrossRef - Farnesoid X Receptor Activation Protects Liver From Ischemia/Reperfusion Injury by Up‐Regulating Small Heterodimer Partner in Kupffer Cells
Dan Jin, Tianfei Lu, Ming Ni, Han Wang, Jiang Zhang, Chenpeng Zhong, Chuan Shen, Jun Hao, Ronald W. Busuttil, Jerzy W. Kupiec‐Weglinski, Jianjun Zhang, Ning Xu, Yuan Zhai
Hepatology Communications.2020; 4(4): 540. CrossRef - Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets
V. Kumar
International Immunopharmacology.2020; 89: 107087. CrossRef - Small Heterodimer Partner Controls the Virus-Mediated Antiviral Immune Response by Targeting CREB-Binding Protein in the Nucleus
Jae-Hoon Kim, Ji-Eun Yoon, Chamilani Nikapitiya, Tae-Hwan Kim, Md Bashir Uddin, Hyun-Cheol Lee, Yong-Hoon Kim, Jung Hwan Hwang, Kiramage Chathuranga, W.A. Gayan Chathuranga, Hueng-Sik Choi, Chul-Joong Kim, Jae U. Jung, Chul-Ho Lee, Jong-Soo Lee
Cell Reports.2019; 27(7): 2105. CrossRef - Glycogen synthase kinase 3β promotes liver innate immune activation by restraining AMP-activated protein kinase activation
Haoming Zhou, Han Wang, Ming Ni, Shi Yue, Yongxiang Xia, Ronald W. Busuttil, Jerzy W. Kupiec-Weglinski, Ling Lu, Xuehao Wang, Yuan Zhai
Journal of Hepatology.2018; 69(1): 99. CrossRef - Cholangiocyte‐derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans
Xiaojiaoyang Li, Runping Liu, Zhiming Huang, Emily C. Gurley, Xuan Wang, Juan Wang, Hongliang He, Hu Yang, Guanhua Lai, Luyong Zhang, Jasmohan S. Bajaj, Melanie White, William M. Pandak, Phillip B. Hylemon, Huiping Zhou
Hepatology.2018; 68(2): 599. CrossRef - Articles inEndocrinology and Metabolismin 2016
Won-Young Lee
Endocrinology and Metabolism.2017; 32(1): 62. CrossRef - Emerging roles of orphan nuclear receptors in regulation of innate immunity
Hyo Sun Jin, Tae Sung Kim, Eun-Kyeong Jo
Archives of Pharmacal Research.2016; 39(11): 1491. CrossRef
 PubReader
PubReader Cite
Cite

