
Search
- Page Path
- HOME > Search
Namgok Lecture 2023
- Diabetes, obesity and metabolism
- Hypothalamic AMP-Activated Protein Kinase as a Whole-Body Energy Sensor and Regulator
- Se Hee Min, Do Kyeong Song, Chan Hee Lee, Eun Roh, Min-Seon Kim
- Endocrinol Metab. 2024;39(1):1-11. Published online February 14, 2024
- DOI: https://doi.org/10.3803/EnM.2024.1922
- 1,932 View
- 76 Download
-
 Abstract
Abstract
 PDF
PDF PubReader
PubReader  ePub
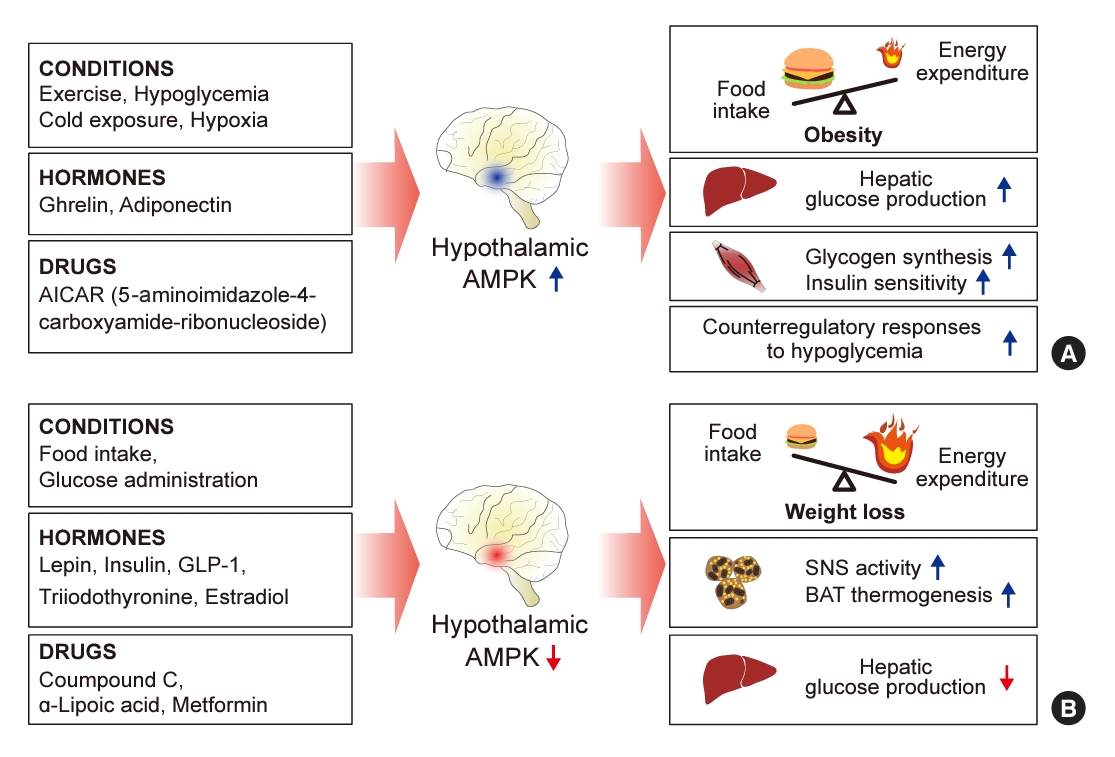
ePub - 5´-Adenosine monophosphate (AMP)-activated protein kinase (AMPK), a cellular energy sensor, is an essential enzyme that helps cells maintain stable energy levels during metabolic stress. The hypothalamus is pivotal in regulating energy balance within the body. Certain neurons in the hypothalamus are sensitive to fluctuations in food availability and energy stores, triggering adaptive responses to preserve systemic energy equilibrium. AMPK, expressed in these hypothalamic neurons, is instrumental in these regulatory processes. Hypothalamic AMPK activity is modulated by key metabolic hormones. Anorexigenic hormones, including leptin, insulin, and glucagon-like peptide 1, suppress hypothalamic AMPK activity, whereas the hunger hormone ghrelin activates it. These hormonal influences on hypothalamic AMPK activity are central to their roles in controlling food consumption and energy expenditure. Additionally, hypothalamic AMPK activity responds to variations in glucose concentrations. It becomes active during hypoglycemia but is deactivated when glucose is introduced directly into the hypothalamus. These shifts in AMPK activity within hypothalamic neurons are critical for maintaining glucose balance. Considering the vital function of hypothalamic AMPK in the regulation of overall energy and glucose balance, developing chemical agents that target the hypothalamus to modulate AMPK activity presents a promising therapeutic approach for metabolic conditions such as obesity and type 2 diabetes mellitus.

Original Articles
- Diabetes, obesity and metabolism
- Inhibition of Sodium-Glucose Cotransporter-2 during Serum Deprivation Increases Hepatic Gluconeogenesis via the AMPK/AKT/FOXO Signaling Pathway
- Jinmi Lee, Seok-Woo Hong, Min-Jeong Kim, Yu-Mi Lim, Sun Joon Moon, Hyemi Kwon, Se Eun Park, Eun-Jung Rhee, Won-Young Lee
- Endocrinol Metab. 2024;39(1):98-108. Published online January 3, 2024
- DOI: https://doi.org/10.3803/EnM.2023.1786

- 1,441 View
- 83 Download
-
Abstract
PDF
 Supplementary MaterialPubReader ePub
Supplementary MaterialPubReader ePub - Background
Sodium-dependent glucose cotransporter 2 (SGLT2) mediates glucose reabsorption in the renal proximal tubules, and SGLT2 inhibitors are used as therapeutic agents for treating type 2 diabetes mellitus. This study aimed to elucidate the effects and mechanisms of SGLT2 inhibition on hepatic glucose metabolism in both serum deprivation and serum supplementation states.
Methods
Huh7 cells were treated with the SGLT2 inhibitors empagliflozin and dapagliflozin to examine the effect of SGLT2 on hepatic glucose uptake. To examine the modulation of glucose metabolism by SGLT2 inhibition under serum deprivation and serum supplementation conditions, HepG2 cells were transfected with SGLT2 small interfering RNA (siRNA), cultured in serum-free Dulbecco’s modified Eagle’s medium for 16 hours, and then cultured in media supplemented with or without 10% fetal bovine serum for 8 hours.
Results
SGLT2 inhibitors dose-dependently decreased hepatic glucose uptake. Serum deprivation increased the expression levels of the gluconeogenesis genes peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α), glucose 6-phosphatase (G6pase), and phosphoenolpyruvate carboxykinase (PEPCK), and their expression levels during serum deprivation were further increased in cells transfected with SGLT2 siRNA. SGLT2 inhibition by siRNA during serum deprivation induces nuclear localization of the transcription factor forkhead box class O 1 (FOXO1), decreases nuclear phosphorylated-AKT (p-AKT), and p-FOXO1 protein expression, and increases phosphorylated-adenosine monophosphate-activated protein kinase (p-AMPK) protein expression. However, treatment with the AMPK inhibitor, compound C, reversed the reduction in the protein expression levels of nuclear p- AKT and p-FOXO1 and decreased the protein expression levels of p-AMPK and PEPCK in cells transfected with SGLT2 siRNA during serum deprivation.
Conclusion
These data show that SGLT2 mediates glucose uptake in hepatocytes and that SGLT2 inhibition during serum deprivation increases gluconeogenesis via the AMPK/AKT/FOXO1 signaling pathway.
- Endocrine Research
- Rebound Feeding in the Wake of Short-Term Suspension of Food Intake Differs in the Presence of Estrous Cycle Peak versus Nadir Levels of Estradiol
- Manita Shakya, Karen P. Briski
- Endocrinol Metab. 2017;32(4):475-484. Published online December 14, 2017
- DOI: https://doi.org/10.3803/EnM.2017.32.4.475
- 3,759 View
- 25 Download
- 2 Web of Science
- 2 Crossref
-
Abstract
PDFPubReader
Background Short-term interruption of feeding is ordinary in modern life but negatively impacts appetite control and body weight. Estradiol (E) imposes long-term inhibitory tonus on food consumption; however, E influence on energy repletion secondary to food deprivation (FD) is unclear. This study investigated the hypothesis that E signal strength regulates hyperphagic responses to FD of varying duration.
Methods Ovariectomized female rats were implanted with E-containing silastic capsules (30 [E-30] or 300 µg [E-300]/mL) to replicate plasma concentrations at cycle nadir versus peak levels.
Results Data show that food intake was increased equally in E-30 and E-300 rats after 12 hours of food deprivation (FD-12); yet, FD of 18 hours (FD-18) amplified refeeding by E-300 versus E-30. Caudal fourth ventricular administration of the 5′-monophosphate-activated protein kinase (AMPK) inhibitor compound C (Cc) did not modify FD-induced hyperphagia in E-30 (regardless of FD interval) or E-300 animals exposed to FD-12, but diminished refeeding after FD-18 in E-300 rats. Cc-reversible hyperglycemia occurred in refed FD-18 groups. Serum insulin was resistant to FD-12 plus refeeding, but was elevated by AMPK-dependent mechanisms in refed E-300 FD-18 rats; equivalent Cc-insensitive decrements in circulating leptin occurred in all FD groups.
Conclusion Current results show that estrous cycle peak, but not baseline, E levels engage hindbrain AMPK signaling to intensify hyperphagia in response to prolongation of FD. Observations of hindbrain AMPK-dependent hyperglycemia, alongside elevated insulin secretion, in refed rats exposed to FD-18 implicate this sensor in insulin resistance mechanisms of glucose partitioning in response to this metabolic imbalance.
-
Citations
Citations to this article as recorded by
- A Framework for Developing Translationally Relevant Animal Models of Stress-Induced Changes in Eating Behavior
Marie François, Olaya Fernández-Gayol, Lori M. Zeltser
Biological Psychiatry.2022; 91(10): 888. CrossRef - Assessing the effects of stress on feeding behaviors in laboratory mice
Marie Francois, Isabella Canal Delgado, Nikolay Shargorodsky, Cheng-Shiun Leu, Lori Zeltser
eLife.2022;[Epub] CrossRef
- A Framework for Developing Translationally Relevant Animal Models of Stress-Induced Changes in Eating Behavior
- Obesity and Metabolism
- Exendin-4 Inhibits the Expression of SEPP1 and Fetuin-A via Improvement of Palmitic Acid-Induced Endoplasmic Reticulum Stress by AMPK
- Jinmi Lee, Seok-Woo Hong, Se Eun Park, Eun-Jung Rhee, Cheol-Young Park, Ki-Won Oh, Sung-Woo Park, Won-Young Lee
- Endocrinol Metab. 2015;30(2):177-184. Published online June 30, 2015
- DOI: https://doi.org/10.3803/EnM.2015.30.2.177
- 4,529 View
- 45 Download
- 12 Web of Science
- 10 Crossref
-
Abstract
PDFPubReader
Background Selenoprotein P (SEPP1) and fetuin-A, both circulating liver-derived glycoproteins, are novel biomarkers for insulin resistance and nonalcoholic fatty liver disease. However, the effect of exendin-4 (Ex-4), a glucagon-like peptide-1 receptor agonist, on the expression of hepatokines, SEPP1, and fetuin-A, is unknown.
Methods The human hepatoma cell line HepG2 was treated with palmitic acid (PA; 0.4 mM) and tunicamycin (tuni; 2ug/ml) with or without exendin-4 (100 nM) for 24 hours. The change in expression of PA-induced SEPP1, fetuin-A, and endoplasmic reticulum (ER) stress markers by exendin-4 treatment were evaluated using quantitative real-time reverse transcription polymerase chain reaction and Western blotting. Transfection of cells with AMP-activated protein kinase (AMPK) small interfering RNA (siRNA) was performed to establish the effect of exendin-4-mediated AMPK in the regulation of SEPP1 and fetuin-A expression.
Results Exendin-4 reduced the expression of SEPP1, fetuin-A, and ER stress markers including PKR-like ER kinase, inositol-requiring kinase 1α, activating transcription factor 6, and C/EBP homologous protein in HepG2 cells. Exendin-4 also reduced the expression of SEPP1 and fetuin-A in cells treated with tunicamycin, an ER stress inducer. In cells treated with the AMPK activator 5-aminoidazole-4-carboxamide ribonucleotide (AICAR), the expression of hepatic SEPP1 and fetuin-A were negatively related by AMPK, which is the target of exendin-4. In addition, exendin-4 treatment did not decrease SEPP1 and fetuin-A expression in cells transfected with AMPK siRNA.
Conclusion These data suggest that exendin-4 can attenuate the expression of hepatic SEPP1 and fetuin-A via improvement of PA-induced ER stress by AMPK.
-
Citations
Citations to this article as recorded by- Maternal Organic Selenium Supplementation Relieves Intestinal Endoplasmic Reticulum Stress in Piglets by Enhancing the Expression of Glutathione Peroxidase 4 and Selenoprotein S
Dajiang Ding, Daolin Mou, Heng Zhu, Xuemei Jiang, Lianqiang Che, Zhengfeng Fang, Shengyu Xu, Yan Lin, Yong Zhuo, Jian Li, Chao Huang, Yuanfeng Zou, Lixia Li, De Wu, Bin Feng
Frontiers in Nutrition.2022;[Epub] CrossRef - Alliin, capsaicin, and gingerol attenuate endoplasmic reticulum stress-induced hepatic steatosis in HepG2 cells and C57BL/6N mice
Ye-Rang Yun, Ji-Eun Lee
Journal of Functional Foods.2022; 95: 105186. CrossRef - PNPLA3 I148M is involved in the variability in anti-NAFLD response to exenatide
Yunzhi Chen, Xuemei Yan, Xiao Xu, Shuhua Yuan, Fen Xu, Hua Liang
Endocrine.2020; 70(3): 517. CrossRef - Green tea extracts reduce leukocyte cell–Derived chemotaxin 2 and selenoprotein P levels in the livers of C57BL/6J mice fed a high-fat diet
Shintaro Onishi, Hidefumi Kitazawa, Shinichi Meguro, Ichiro Tokimitsu
Bioscience, Biotechnology, and Biochemistry.2018; 82(9): 1568. CrossRef - Melatonin improves insulin resistance and hepatic steatosis through attenuation of alpha‐2‐HS‐glycoprotein
Jee‐In Heo, Dae Wui Yoon, Ji Hee Yu, Nam Hoon Kim, Hye Jin Yoo, Ji A. Seo, Sin Gon Kim, Kyung Mook Choi, Sei Hyun Baik, Dong Seop Choi, Nan Hee Kim
Journal of Pineal Research.2018;[Epub] CrossRef - Palmitic acid induces ceramide accumulation, mitochondrial protein hyperacetylation, and mitochondrial dysfunction in porcine oocytes†
Nobuhiko Itami, Koumei Shirasuna, Takehito Kuwayama, Hisataka Iwata
Biology of Reproduction.2018; 98(5): 644. CrossRef - Astragaloside IV attenuates free fatty acid-induced ER stress and lipid accumulation in hepatocytes via AMPK activation
Bing Zhou, Dan-li Zhou, Xiao-hong Wei, Rong-yu Zhong, Jie Xu, Liao Sun
Acta Pharmacologica Sinica.2017; 38(7): 998. CrossRef - New Potential Targets of Glucagon-Like Peptide 1 Receptor Agonists in Pancreatic β-Cells and Hepatocytes
Won-Young Lee
Endocrinology and Metabolism.2017; 32(1): 1. CrossRef - Selenoprotein P neutralizes lipopolysaccharide and participates in hepatic cell endoplasmic reticulum stress response
Yongzhong Zhao, Shuvojit Banerjee, Ping Huang, Xinning Wang, Candece L. Gladson, Warren D. Heston, Charles B. Foster
FEBS Letters.2016; 590(24): 4519. CrossRef - Novel phenotypes of prediabetes?
Hans-Ulrich Häring
Diabetologia.2016; 59(9): 1806. CrossRef
- Maternal Organic Selenium Supplementation Relieves Intestinal Endoplasmic Reticulum Stress in Piglets by Enhancing the Expression of Glutathione Peroxidase 4 and Selenoprotein S
- Obesity and Metabolism
- Activation of AMP-Activated Protein Kinase Attenuates Tumor Necrosis Factor-α-Induced Lipolysis via Protection of Perilipin in 3T3-L1 Adipocytes
- Seok-Woo Hong, Jinmi Lee, Se Eun Park, Eun-Jung Rhee, Cheol-Young Park, Ki-Won Oh, Sung-Woo Park, Won-Young Lee
- Endocrinol Metab. 2014;29(4):553-560. Published online December 29, 2014
- DOI: https://doi.org/10.3803/EnM.2014.29.4.553
- 3,427 View
- 26 Download
- 12 Web of Science
- 10 Crossref
-
Abstract
PDFPubReader
Background Tumor necrosis factor (TNF)-α and AMP-activated protein kinase (AMPK) are known to stimulate and repress lipolysis in adipocytes, respectively; however, the mechanisms regulating these processes have not been completely elucidated.
Methods The key factors and mechanism of action of TNF-α and AMPK in lipolysis were investigated by evaluating perilipin expression and activity of protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 α (eIF2α) by Western blot and an immunofluorescence assay in 24-hour TNF-α-treated 3T3-L1 adipocytes with artificial manipulation of AMPK activation.
Results Enhancement of AMPK activity by the addition of activator minoimidazole carboxamide ribonucleotide (AICAR) suppressed TNF-α-induced lipolysis, whereas the addition of compound C, an inhibitor of AMPK phosphorylation, enhanced lipolysis. Perilipin, a lipid droplet-associated protein, was decreased by TNF-α and recovered following treatment with AICAR, showing a correlation with the antilipolytic effect of AICAR. Significant activation of PERK/eIF2α, a component of the unfolded protein response signaling pathway, was observed in TNF-α or vesicle-treated 3T3-L1 adipocytes. The antilipolytic effect and recovery of perilipin expression by AICAR in TNF-α-treated 3T3-L1 adipocytes were significantly diminished by treatment with 2-aminopurine, a specific inhibitor of eIF2α.
Conclusion These data indicated that AICAR-induced AMPK activation attenuates TNF-α-induced lipolysis via preservation of perilipin in 3T3-L1 adipocytes. In addition, PERK/eIF2α activity is a novel mechanism of the anti-lipolytic effect of AICAR.
-
Citations
Citations to this article as recorded by- Dysregulation of Lipid Droplet Protein Expression in Adipose Tissues and Association with Metabolic Risk Factors in Adult Females with Obesity and Type 2 Diabetes
Chan Yoon Park, Donguk Kim, Min Kyeong Seo, Jimin Kim, Han Choe, Jong-Hyeok Kim, Joon Pio Hong, Yeon Ji Lee, Yoonseok Heo, Hwa Jung Kim, Hye Soon Park, Yeon Jin Jang
The Journal of Nutrition.2023; 153(3): 691. CrossRef - Tschimganidine reduces lipid accumulation through AMPK activation and alleviates high-fat diet-induced metabolic diseases
Min-Seon Hwang, Jung-Hwan Baek, Jun-Kyu Song, In Hye Lee, Kyung-Hee Chun
BMB Reports.2023; 56(4): 246. CrossRef - Acetate stimulates lipogenesis via AMPKα signaling in rabbit adipose-derived stem cells
Lei Liu, Chunyan Fu, Yongxu Liu, Fuchang Li
General and Comparative Endocrinology.2021; 303: 113715. CrossRef - Docosahexaenoic acid-enriched phospholipids and eicosapentaenoic acid-enriched phospholipids inhibit tumor necrosis factor-alpha-induced lipolysis in 3T3-L1 adipocytes by activating sirtuin 1 pathways
Yu-Hong Yang, Yi-Ming Hao, Xiao-Fang Liu, Xiang Gao, Bao-Zhen Wang, Koretaro Takahashi, Lei Du
Food & Function.2021; 12(11): 4783. CrossRef - Role of the AMPK/ACC Signaling Pathway in TRPP2-Mediated Head and Neck Cancer Cell Proliferation
Kun Li, Lei Chen, Zhangying Lin, Junwei Zhu, Yang Fang, Juan Du, Bing Shen, Kaile Wu, Yehai Liu, Gianmarco Saponaro
BioMed Research International.2020; 2020: 1. CrossRef - GLUT12 and adipose tissue: Expression, regulation and its relation with obesity in mice
Eva Gil‐Iturbe, José Miguel Arbones‐Mainar, María J. Moreno‐Aliaga, María Pilar Lostao
Acta Physiologica.2019;[Epub] CrossRef - Bilobalide Suppresses Adipogenesis in 3T3-L1 Adipocytes via the AMPK Signaling Pathway
Su Bu, Chun Ying Yuan, Quan Xue, Ying Chen, Fuliang Cao
Molecules.2019; 24(19): 3503. CrossRef - Sulforaphane induces adipocyte browning and promotes glucose and lipid utilization
Hui Q. Zhang, Shi Y. Chen, An S. Wang, An J. Yao, Jian F. Fu, Jin S. Zhao, Fen Chen, Zu Q. Zou, Xiao H. Zhang, Yu J. Shan, Yong P. Bao
Molecular Nutrition & Food Research.2016; 60(10): 2185. CrossRef - Fyn phosphorylates AMPK to inhibit AMPK activity and AMP-dependent activation of autophagy
Eijiro Yamada, Shuichi Okada, Claire C. Bastie, Manu Vatish, Yasuyo Nakajima, Ryo Shibusawa, Atsushi Ozawa, Jeffrey E. Pessin, Masanobu Yamada
Oncotarget.2016; 7(46): 74612. CrossRef - Articles in 'Endocrinology and Metabolism' in 2014
Won-Young Lee
Endocrinology and Metabolism.2015; 30(1): 47. CrossRef
- Dysregulation of Lipid Droplet Protein Expression in Adipose Tissues and Association with Metabolic Risk Factors in Adult Females with Obesity and Type 2 Diabetes
- Obesity and Metabolism
- Vav3, a GEF for RhoA, Plays a Critical Role under High Glucose Conditions
- Jie Sha, Jungsik Na, Jung Ok Lee, Nami Kim, Soo Kyung Lee, Ji Hae Kim, Ji Wook Moon, Su Jin Kim, Hye Jeong Lee, Jong-Il Choi, Sun Hwa Park, Hyeon Soo Kim
- Endocrinol Metab. 2014;29(3):363-370. Published online September 25, 2014
- DOI: https://doi.org/10.3803/EnM.2014.29.3.363
- 3,743 View
- 39 Download
- 6 Web of Science
- 6 Crossref
-
Abstract
PDFPubReader
Background The role of small GTPase molecules is poorly understood under high glucose conditions.
Methods We analyzed the expression pattern of Vav3 in skeletal muscle C2C12 cells under high glucose culture condition with reverse transcription-polymerase chain reaction and Western blot analysis. We also measured glucose uptake using isotope-labelled glucose.
Results We showed that expression of Vav3 (a guanine nucleotide exchange factor for RhoA) increased. mRNA and protein levels in skeletal muscle C2C12 cells under high glucose conditions. The AMP-activated protein kinase (AMPK) activator AMPK agonist 5-aminoimidazole-4-carboxy-amide-1-d-ribofuranoside (AICAR) suppressed high glucose-induced Vav3 induction. In addition, exposure of cells to high glucose concentration increased the phosphorylation of PAK-1, a molecule downstream of RhoA. The phosphorylation of paxillin, a downstream molecule of PAK-1, was also increased by exposure to high glucose. Phosphorylation of these molecules was not observed in the presence of AICAR, indicating that AMPK is involved in the RhoA signal pathway under high glucose conditions. Knock down of Vav3 enhances metformin-mediated glucose uptake. Inhibition of AMPK blocked the increases of Vav3 knock down-induced glucose uptake. Metformin-mediated Glut4 translocation was also increased by Vav3 knock-down, suggesting that Vav3 is involved in metformin-mediated glucose uptake.
Conclusion These results demonstrate that Vav3 is involved in the process of metformin-mediated glucose regulation.
-
Citations
Citations to this article as recorded by- Peripheral origin exosomal microRNAs aggravate glymphatic system dysfunction in diabetic cognitive impairment
Lin Zhang, Dongna Li, Pengrong Yi, Jiangwei Shi, Mengqing Guo, Qingsheng Yin, Dingbin Liu, Pengwei Zhuang, Yanjun Zhang
Acta Pharmaceutica Sinica B.2023; 13(7): 2817. CrossRef - A current overview of RhoA, RhoB, and RhoC functions in vascular biology and pathology
Robert Eckenstaler, Michael Hauke, Ralf A. Benndorf
Biochemical Pharmacology.2022; 206: 115321. CrossRef - Rho Family GTPases and Rho GEFs in Glucose Homeostasis
Polly A. Machin, Elpida Tsonou, David C. Hornigold, Heidi C. E. Welch
Cells.2021; 10(4): 915. CrossRef - Pharmacological Modulators of Small GTPases of Rho Family in Neurodegenerative Diseases
William Guiler, Addison Koehler, Christi Boykin, Qun Lu
Frontiers in Cellular Neuroscience.2021;[Epub] CrossRef - Association of VAV2 and VAV3 polymorphisms with cardiovascular risk factors
Nuria Perretta-Tejedor, Javier Fernández-Mateos, Luis García-Ortiz, Manuel A. Gómez-Marcos, José I. Recio-Rodríguez, Cristina Agudo-Conde, Emiliano Rodriguez-Sánchez, Ana I. Morales, Francisco J. López-Hernández, José M. López-Novoa, Rogelio González-Sarm
Scientific Reports.2017;[Epub] CrossRef - Articles in 'Endocrinology and Metabolism' in 2014
Won-Young Lee
Endocrinology and Metabolism.2015; 30(1): 47. CrossRef
- Peripheral origin exosomal microRNAs aggravate glymphatic system dysfunction in diabetic cognitive impairment
 First
First Prev
Prev


