Articles
- Page Path
- HOME > Endocrinol Metab > Volume 37(1); 2022 > Article
-
Review ArticleDiabetes, Obesity and Metabolism Homeostatic Regulation of Glucose Metabolism by the Central Nervous System
-
Jong Han Choi1
 , Min-Seon Kim2,3
, Min-Seon Kim2,3 -
Endocrinology and Metabolism 2022;37(1):9-25.
DOI: https://doi.org/10.3803/EnM.2021.1364
Published online: February 28, 2022
1Division of Endocrinology and Metabolism, Konkuk University Medical Center, Seoul, Korea
2Division of Endocrinology and Metabolism, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
3Appeptite Regulation Laboratory, Asan Institute for Life Sciences, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding author: Min-Seon Kim. Division of Endocrinology and Metabolism, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea, Tel: +82-2-3010-3245, Fax: +82-2-3010-6962, E-mail: mskim@amc.seoul.kr
Copyright © 2022 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- THE NEURONS AND NEURAL CIRCUITS CONTROLLING GLUCOSE HOMEOSTASIS
- GLUCOSE-SENSING MECHANISMS IN THE BRAIN
- CONTRIBUTION OF CENTRAL INSULIN SIGNALING TO SYSTEMIC GLUCOSE METABOLISM
- OTHER HORMONAL SIGNALING MECHANISMS INVOLVED IN BRAIN REGULATION OF GLUCOSE METABOLISM
- OBESITY-INDUCED HYPOTHALAMIC INFLAMMATION AND ITS POSSIBLE IMPACTS ON GLUCOSE HOMEOSTASIS
- CONCLUSIONS
- Article information
- References
ABSTRACT
- Evidence for involvement of the central nervous system (CNS) in the regulation of glucose metabolism dates back to the 19th century, although the majority of the research on glucose metabolism has focused on the peripheral metabolic organs. Due to recent advances in neuroscience, it has now become clear that the CNS is indeed vital for maintaining glucose homeostasis. To achieve normoglycemia, specific populations of neurons and glia in the hypothalamus sense changes in the blood concentrations of glucose and of glucoregulatory hormones such as insulin, leptin, glucagon-like peptide 1, and glucagon. This information is integrated and transmitted to other areas of the brain where it eventually modulates various processes in glucose metabolism (i.e., hepatic glucose production, glucose uptake in the brown adipose tissue and skeletal muscle, pancreatic insulin and glucagon secretion, renal glucose reabsorption, etc.). Errors in these processes lead to hyper- or hypoglycemia. We here review the current understanding of the brain regulation of glucose metabolism.
- Glucose is a primary energy source in mammals including humans and it is thus essential for higher organisms to monitor and control glucose availability within strict physiological ranges. Mammals have a dedicated sensor and effector system for glucose homeostasis in the brain as neurons exclusively use glucose as their energy source, and the brain is the most active glucose consumer. Sensing the blood glucose levels and preventing hypoglycemia is therefore crucial for brain functions such as cognition, memory, and motor functions. The involvement of the brain in glucose regulation was first described in 1854 by French physiologist Claude Bernard who observed a dramatic rise in blood glucose levels when the floor of the fourth ventricle of a rabbit brain was stimulated [1]. In the early 1950s, the French-American scientist Jean Mayer [2] reported that the brain detects glucose fluctuations. In the mid-1960s, experiments using electrophysiological recordings led to the discovery of glucose-sensing neurons and demonstrated their role in maintaining normoglycemia [3–5]. Many studies have since shown that various regions of the hypothalamus and brainstem are involved in the central regulation of glucose metabolism through their interactions with peripheral metabolic organs [6], as depicted in Fig. 1. We here discuss the mechanisms by which the brain regulates glucose metabolism.
INTRODUCTION
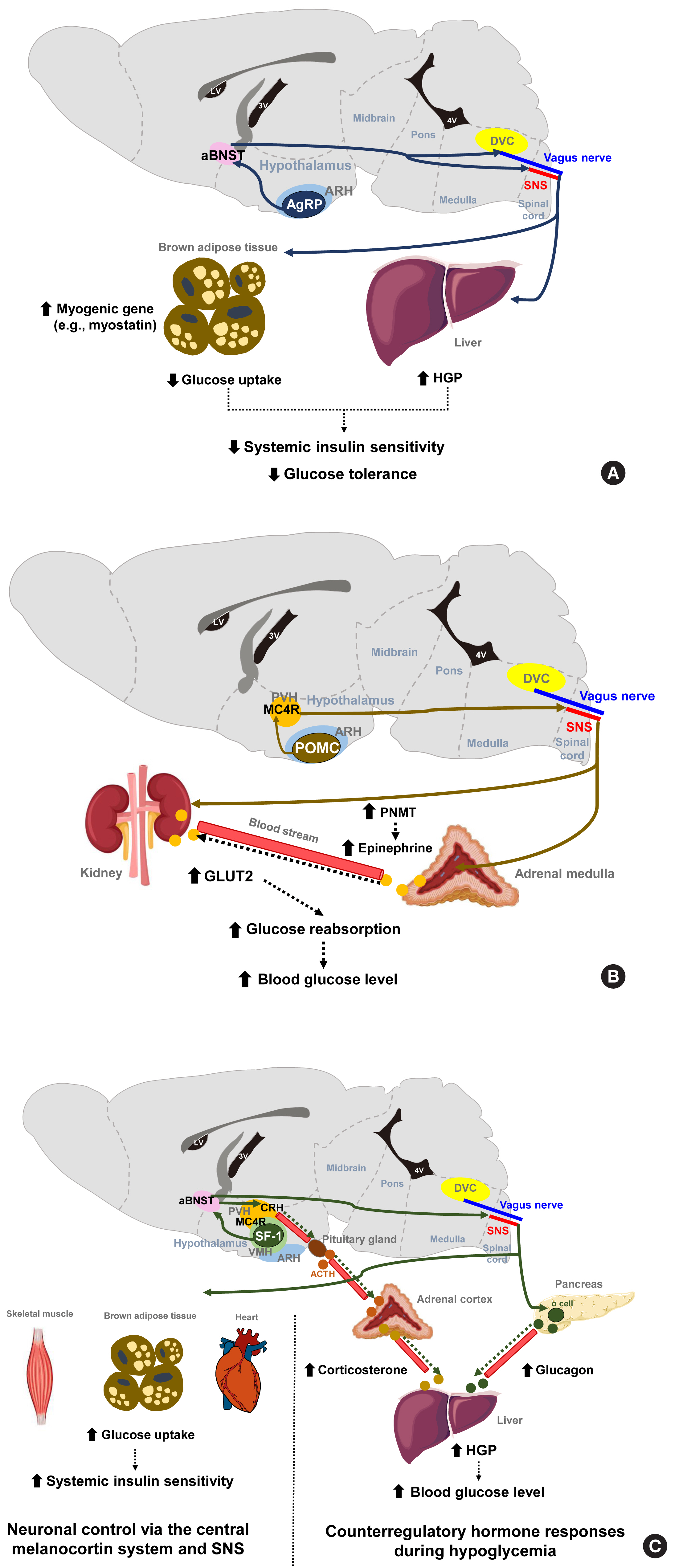
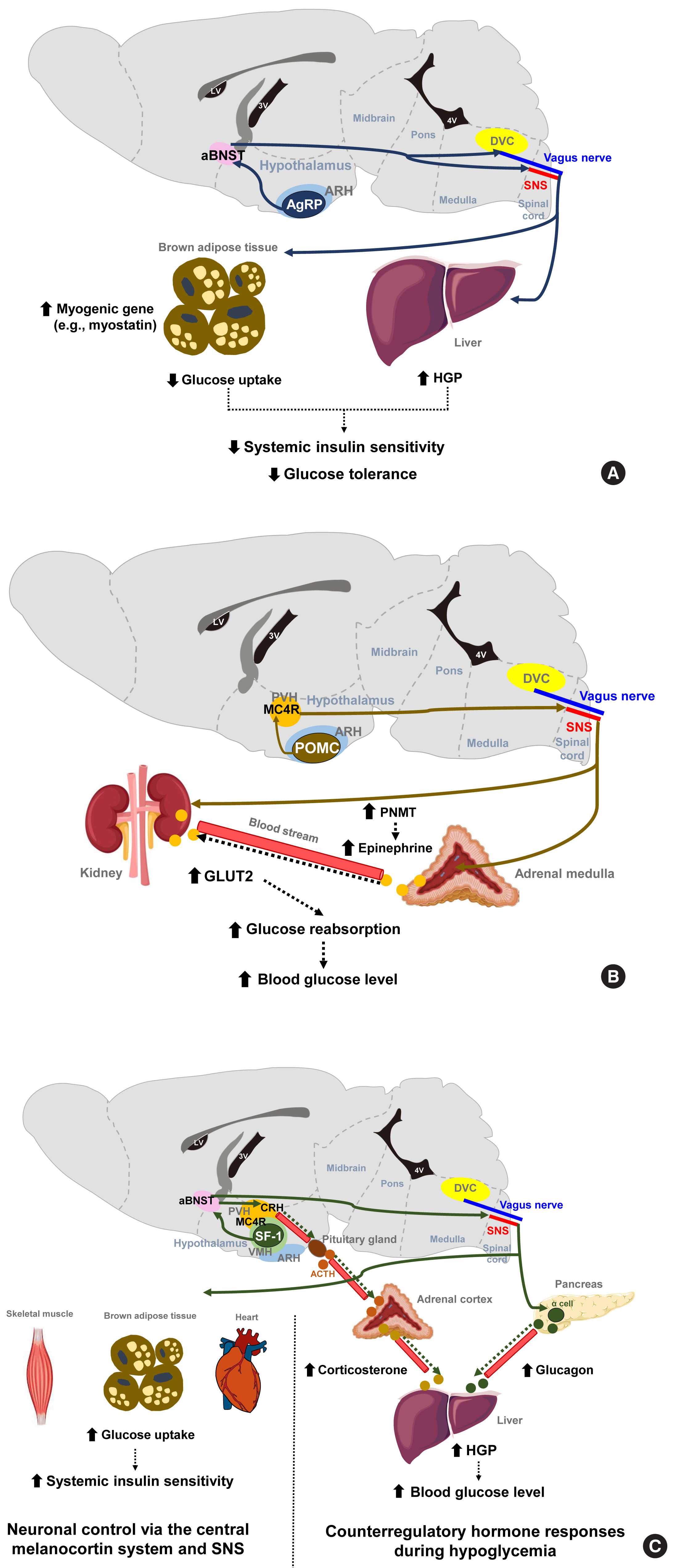
- The hypothalamus has a central function in the neural networks that regulate glucose metabolism. The arcuate nucleus of the hypothalamus (ARH) is located at the bottom of the third ventricle and two neuronal populations in the ARH are considered to be critical for the brain’s regulation of energy balance and glucose metabolism. One of these groups of neurons secretes α-melanocyte stimulating hormone (α-MSH) derived from the precursor peptide proopiomelanocortin (POMC) and the other co-secretes Agouti-related protein (AgRP) and neuropeptide Y (NPY). Regarding energy metabolism, POMC neurons have catabolic actions that are largely through its agonistic actions on the melanocortin-4 receptor (MC4R), whereas AgRP/NPY neurons have anabolic effects that are partly mediated via MC4R antagonism. Recently developed chemogenetic and optogenetic technologies have enabled the control of neuronal activity in specific neuronal populations and neural circuits in live animals in order to study their roles in the control of glucose metabolism.
- The stimulation of AgRP neuronal activity using optogenetic and chemogenetic tools decreases the systemic insulin sensitivity and glucose tolerance levels [7]. This effect is partly mediated through the induction of myogenic genes (such as myostatin) and the suppression of glucose uptake in the brown adipose tissue (BAT) and is independent of MC4R antagonism. In addition, AgRP neurons are shown to regulate hepatic glucose production (HGP) [8]. Notably, the neural circuitry involving ARH AgRP neuronal projections to the ventro-lateral part of anterior bed nucleus of the stria terminalis (aBNST) specifically conveys the effects of AgRP stimulation on glucose metabolism (Fig. 1A) [7], whereas their axonal projections to the paraventricular hypothalamus (PVH), the lateral hypothalamus (LH) area, and the dorsomedial part of aBNST control feeding [7].
- POMC neurons are shown to mediate the glucose-lowering effects of leptin [9]. In contrast, the acute chemogenetic inhibition of POMC neuronal activity lowers the glycemic level independently of food intake [10]. Moreover, mice that have received ARH-specific POMC ablation show an improved glucose tolerance due to enhanced renal glucose excretion and despite the concomitant induction of obesity and insulin resistance [11]. That same study and another prior investigation reported that ARH POMC neurons promote renal glucose reabsorption via central MC4R signaling, renal sympathetic innervation, and the upregulation of renal expression of type 2 glucose transporter (GLUT2) (Fig. 1B) [11,12].
- The ventromedial hypothalamus (VMH) is important for counterregulatory hormone responses and in the recovery from hypoglycemia [13]. When the activity of VMH steroidogenic factor-1 expressing (SF-1) neurons is optogenetically inhibited, animals fail to recover from insulin-induced hypoglycemia owing to impaired counterregulatory hormone responses (reduced glucagon and corticosterone secretion) and decreased HGP [14]. Similar to AgRP neurons, the neural pathway to the aBNST is critical for the regulation of counterregulatory hormone responses by SF-1 neurons [14]. On the other hand, SF-1 neurons also regulate insulin sensitivity in peripheral metabolic organs. The chemical and genetic activation of VMH SF-1 neurons increases systemic insulin sensitivity and glucose uptake in red skeletal muscle, heart and BAT during a hyperinsulinemic-euglycemic clamp [15]. These effects are mediated by central melanocortin signaling and the sympathetic nervous system (Fig. 1C) [16,17]. Consistently, the SF-1 neuron-restricted reexpression of uncoupling protein 2 (UCP2) in generalized UCP2 null mice was reported to activate systemic insulin sensitivity and glucose tolerance by enhancing insulin signaling in the liver, skeletal muscle, and BAT [18].
- The LH is known as a feeding center, and its effects are mediated by the inhibitory input from the BNST to the LH glutamatergic neurons [19]. Specific MC4R reexpression in the LH of generalized MC4R-null mice improves glucose tolerance and glycemia in normal chow diet- and high-fat diet (HFD)-fed mice. This glucose-lowering effect of LH MC4R signaling is mediated by upregulation of glucose uptake and thermogenesis in the BAT through the control of BAT sympathetic nerve activity [20]. On the other hand, LH orexin-producing neurons are activated by sweet foods, and they regulate skeletal muscle glucose uptake through VMH orexin receptor-expressing neurons and the sympathetic nervous system [21]. The preoptic area (POA) in the anterior hypothalamus regulates BAT thermogenesis [22]. Given the close link between BAT thermogenesis and glucose tolerance, the involvement of POA neurons in glycemic control needs to be tested.
- The dorsal vagal complex (DVC) in the brainstem is crucial for sensing glucose fluctuations and regulating glycemic levels and food intake [23,24]. The DVC includes the nucleus tractus solitarius (NTS), the dorsal motor nucleus of the vagus (DMV), and the area postrema (AP). Meal-related signals from the gastrointestinal tract are relayed to the NTS and the AP through the sensory vagus nerve [25]. Like hypothalamic neurons, NTS neurons produce appetite-regulating peptides such as NPY, POMC, and glucagon-like peptide 1 (GLP-1) [26,27]. The NTS POMC neurons are involved in the short-term regulation of feeding, but the destruction of these neurons neither alters body weight nor glucose metabolism [28]. MC4R is also expressed in the brainstem neurons. Deletion of MC4R in cholinergic sympathetic and parasympathetic preganglionic neurons using Chat-Cre mice impairs glucose homeostasis [29], whereas its reactivation in MC4R null mice attenuates hyperglycemia and insulin resistance through improved hepatic insulin sensitivity. These findings indicate the involvement of brainstem MC4R signaling in the central regulation of glucose metabolism [30]. NTS GABAergic neurons sense hypoglycemia and then help recover from hypoglycemia through DMV projections and the parasympathetic stimulation of glucagon secretion [31]. Conversely, the NTS receives gut-derived, energy-intake signals, which it then transfers to the hypothalamus via the parabrachial nucleus (PBN) (long-loop) or to the DMV (short-loop) to generate satiety [32].
THE NEURONS AND NEURAL CIRCUITS CONTROLLING GLUCOSE HOMEOSTASIS
- In the central nervous system (CNS) regulation of glucose metabolism, glucose by itself acts as an important afferent signal to the brain. In this signaling mechanism, blood glucose enters the brain across the blood-brain barrier (BBB), which is mediated by insulin−independent and high-affinity glucose transporter type 1 and 3 (GLUT1, GLUT3) expressed on the endothelial cells. This enables efficient glucose transport into the brain in the normoglycemic range in an insulin-independent manner [33].
- Neuronal glucose-sensing
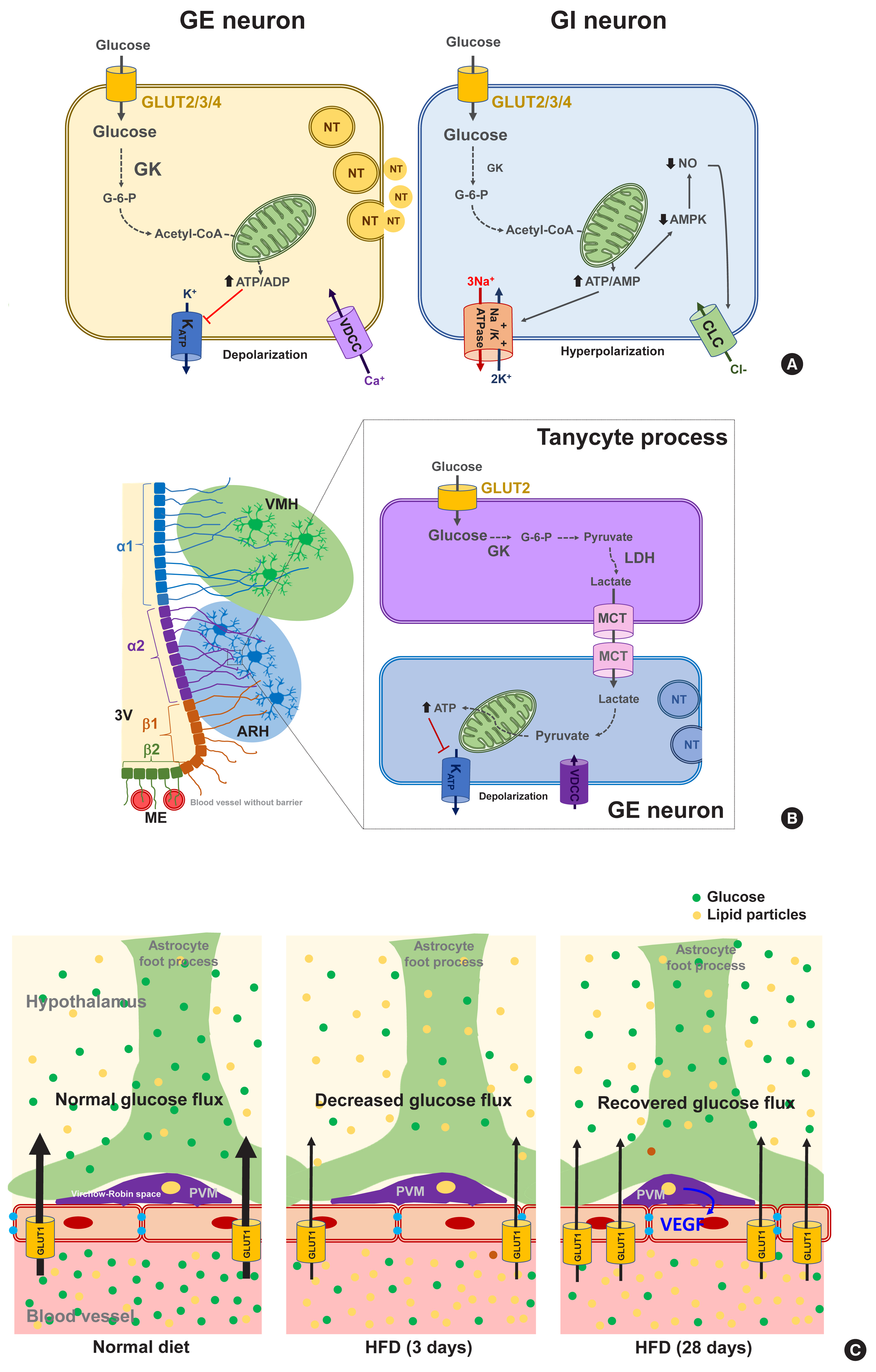
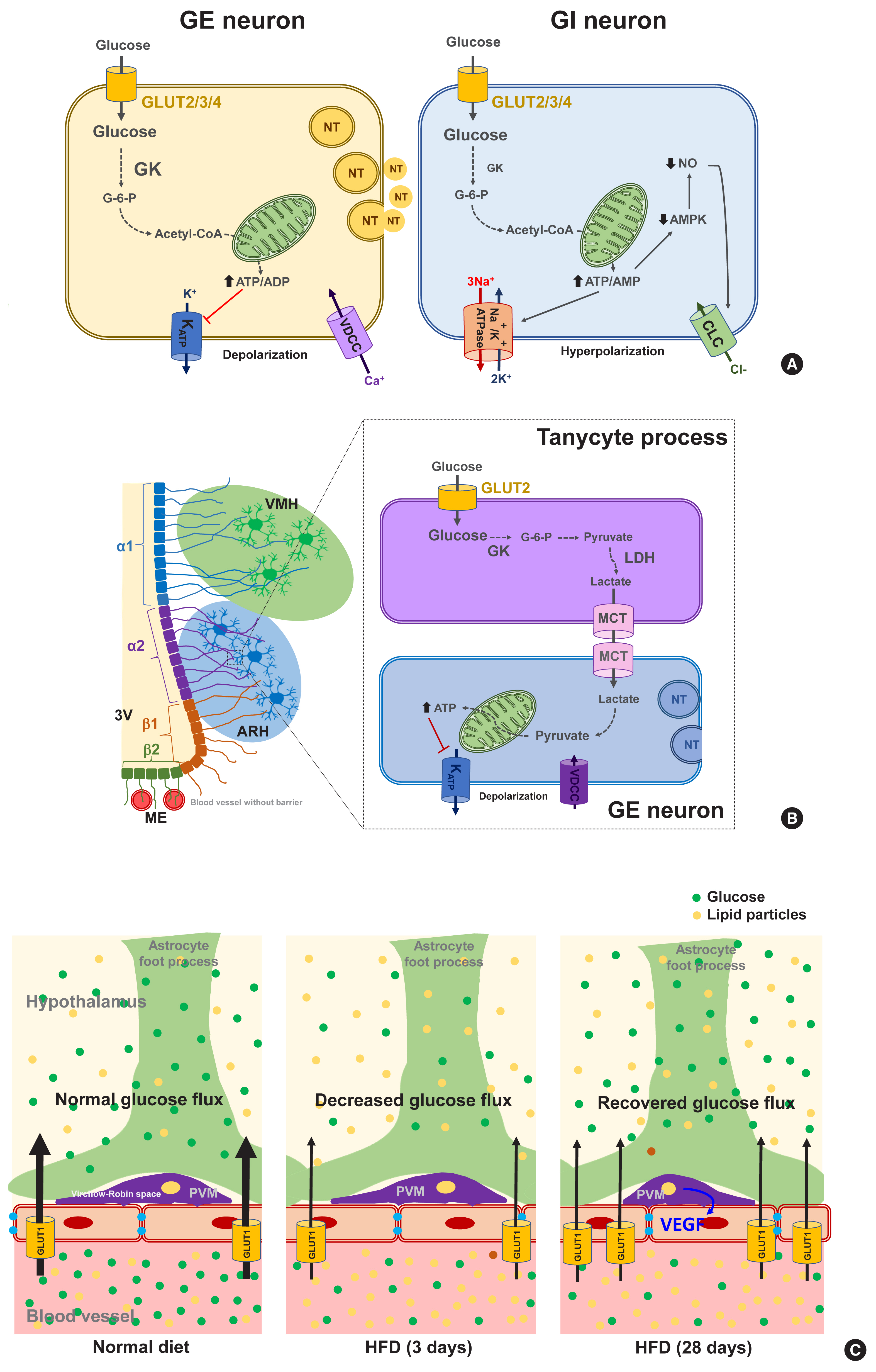
- Glucose-sensing neurons are those that can alter their excitability in response to changes in the extracellular glucose levels and can be categorized as glucose-excitatory (GE) and glucose-inhibitory (GI) neurons [34]. GE neurons are excited when the extracellular glucose level rises and vice versa [35]. The molecular basis of glucose-sensing in GE neurons is similar to the classical glucose-sensing mechanism in pancreatic β-cells (Fig. 2A, left panel), although these pathways are not yet fully understood. In GE neurons, glucose enters the neurons via GLUT2, 3, and 4. Upon uptake, glucose is converted to glucose-6-phosphate by glucokinase (GK) and then undergoes glycolysis and mitochondrial oxidation, resulting in adenosine triphosphate (ATP) production. An increased cellular ATP/adenosine diphosphate (ADP) ratio leads to a number of sequential events, i.e., the closure of ATP-dependent potassium (KATP) channels, depolarization of the membrane potential, Ca2+ influx through voltage-dependent calcium channels, and finally the stimulation of neuronal activity and neurotransmitter release [36]. During glucose-sensing by GE neurons, GK activity is essential and is dependent on cellular glucose levels owing to its low affinity for glucose [34]. A wide body of data has illustrated the roles of hypothalamic GLUTs, GK, and KATP channels in glucose-sensing and glycemic control by the CNS [37–41]. For instance, the genetic deletion of Kir6.2, a subunit of the KATP channel, in ARH POMC neurons reduces glucose tolerance [42]. Acute electromagnetic stimulation of GK-expressing neurons in the VMH increases the blood glucose levels by increasing HGP and glucagon levels. In turn, suppressing these neuronal activities curbs the counterregulatory responses to hypoglycemia through opposing actions [43].
- In contrast to GE neurons, GI neurons are activated when the extracellular glucose level decreases. Relatively little is known however about the underlying mechanism of this. GI neurons also express the same GLUTs and GK as those of GE neurons [34]. However, GK appears to be less essential for glucose-sensing in GI neurons [44]. As a possible mechanism, glucose increases the ATP/adenosine monophosphate (AMP) ratio, which stimulates the Na+/K+-ATPase pump and induces a hyperpolarizing current [45]. Alternatively, glucose may cause the opening of Cl− channels through mechanisms involving adenosine monophosphate-activated protein kinase (AMPK) and nitric oxide (NO), which leads to plasma membrane hyperpolarization (Fig. 2A, right panel) [35,46].
- Glucose-sensing neurons are mainly distributed in the hypothalamic, corticolimbic, and brainstem areas, also known as critical body weight control areas. As the ARH is adjacent to the circumventricular organ median eminence (ME), ARH neurons are the first to sense the changes in blood glucose. Many studies have indicated that POMC neurons are GE, and about 40% of AgRP/NPY neurons are GI [34]. GE neurons are also found in the VMH and the PVH, which are critically involved in satiety generation [47]. In contrast, GI neurons are distributed in the LH, which has long been known as a hunger center [35,48].
- Both GE and GI neurons are also present in the brainstem areas, including the DMV, NTS, and PBN [49–51], whereas the PBN mostly contains GI neurons [52], which is consistent with its role in the adaptative responses to hypoglycemia. In further support of these findings, the local induction of glucoprivation in these aforementioned brainstem areas using a 5-Thio-D-glucose injection triggers hyperglycemic and hyperphagic responses in rats [53]. Glucose-sensing neurons are also found in the limbic regions related to the rewarding such as the nucleus accumbens (NAc) and amygdala [54]. The NAc and amygdala contain both GE and GI neurons. The reasons why neurons in the reward system need to sense glucose remain to be resolved.
- Non-neuronal glucose-sensing
- The mammalian brain comprises both neurons and non-neuronal cells (astrocytes, microglia, oligodendrocytes, nerve/glial antigen 2 [NG2] glia, endothelial cells, pericytes, ependymal cells, and tanycytes). Recent evidence indicates an involvement of non-neuronal cells in brain glucose-sensing. Astrocytes are abundant glial cells that support neuronal functions in many ways. As astrocytes are located at the interface between blood vessels and neurons, they are in a prime location for controlling the glucose flux into the CNS. The foot processes of astrocytes that ensheath the brain microvessels, highly express GLUT1. Blood glucose enters the astrocytes mainly through GLUT1 and then is oxidized in the mitochondria, stored as glycogen, or converted to lactate and secreted. Lactate is an alternative energy source for neurons. Indeed, neurons are believed to be metabolically and functionally coupled with astrocytes through lactate, which is known as the astrocyte-neuron lactate shuttle hypothesis [55].
- Interestingly, persistent hyperglycemia in streptozotocin-induced diabetic rats was reported to reduce the astroglial GLUT1 expression level, and to impair elevation in the hypothalamic glucose concentrations, and HGP suppression after glucose loading [56]. These findings suggest that GLUT1-mediated glial glucose uptake is important for brain glucose-sensing and for the regulation of glucose metabolism. On the other hand, a generalized depletion of GLUT2 is known to impair glucagon secretion during hypoglycemia, and to be rescued by the selective reexpression of GLUT2 in glial fibrillary acidic protein (GFAP)-expressing astroglial cells, but not in neurons [38]. This evidence indicates the critical role of astroglial GLUT2 in sensing hypoglycemia and regulating glucagon secretion.
- Tanycytes are specialized ependymal cells surrounding the third cerebroventricle [57] and have four subtypes: α1, α2, β1, and β2. They are polarized cells as their cell body faces the cerebroventricular side, and their long processes project and contact the neurons in the VMH (α1) and ARH (α2 and β1), or the fenestrated blood vessels in the ME (β2) [58,59]. Thanks to topological characteristics, the tanycytes mediate material transport between the cerebroventricle and the hypothalamic parenchyma (α1, α2, β1) and between the cerebroventricle and blood vessels (β2). For instance, tanycytes have been shown to mediate leptin transport from the blood to the hypothalamus [60]. These cells also express the glucose-sensing molecules GLUT2, GK, and KATP channels [61] and may sense glucose in the cerebrospinal fluid (CSF) which is derived from the blood across the blood–CSF barrier. As indicated in Fig. 2B, tanycytes may play a role in glucose uptake from the CSF via GLUT2 and then release lactate through monocarboxylate transporter. Tanycyte-derived lactate may activate or inhibit the neuronal electrical activity that contacts the processes of these cells, as demonstrated in vitro in a prior study [62]. The selective destruction of tanycytes through the intracerebroventricular injection of alloxan inhibits the counterregulatory responses to hypoglycemia without damaging ARH neurons [63]. These findings support the involvement of tanycytes in hypothalamic glucose-sensing [64], as depicted in Fig. 2B.
- Perivascular macrophages (PVMs) are located in the Virchow-Robin space, a fluid−filled region that surrounds the small arteries in the brain. Given the location of this space at the interface of blood vessels and the brain parenchyma, these cells may sense and regulate glucose flux from the circulation to the brain. Under conditions of a reduced glucose flux, i.e., HFD consumption, PVMs secrete vascular endothelial growth factor (VEGF) which, in turn, acts on the endothelial cells to restore the glucose flux by increasing GLUT1 expression (Fig. 2C) [65]. This mechanism appears to be critical for adequate glucose supply to the CNS neurons and maintenance of cognitive functions in HFD-fed conditions. However, whether this may also be important for controlling glucose metabolism has not been tested yet. Microglia are macrophage-like cells that are evenly distributed throughout the brain parenchyma and execute multiple homeostatic roles and innate immune functions. A recent study has shown that microglia, especially those in proximity to the glucose-sensing NPY neurons, are activated during hypoglycemia. Inhibiting microglial activation using minocycline enhances the counterregulatory hormone responses to hypoglycemia [66], and thus microglial activation around NPY neurons may play a role in the hypoglycemia unawareness induced by repeated exposure to hypoglycemia. These findings imply that microglia may sense fluctuations in nutrient availability and regulate neuronal adaptive responses. Evidence for the contribution of hypothalamic immune cells to glucose homeostasis is beginning to be reported. Therefore, future studies are necessary to reveal how hypothalamic immune cells affect glucose metabolism.
GLUCOSE-SENSING MECHANISMS IN THE BRAIN
- Insulin is the most important hormone in glucose regulation and is generally thought to regulate glucose metabolism via its direct actions on peripheral metabolic organs. However, insulin receptor (IR) and its downstream signaling molecules, insulin receptor substrate 1 and 2 (IRS1/IRS2), are expressed in brain regions including the hypothalamus [67]. Moreover, neuron-specific IR knockout (NIRKO) mice exhibit mild obesity and insulin resistance [68]. Brain-specific IRS2 knockout mice also display overweight, hyperinsulinemia, and glucose intolerance [69]. These findings support the notion that insulin also regulates glucose metabolism and body weight through its actions in the brain.
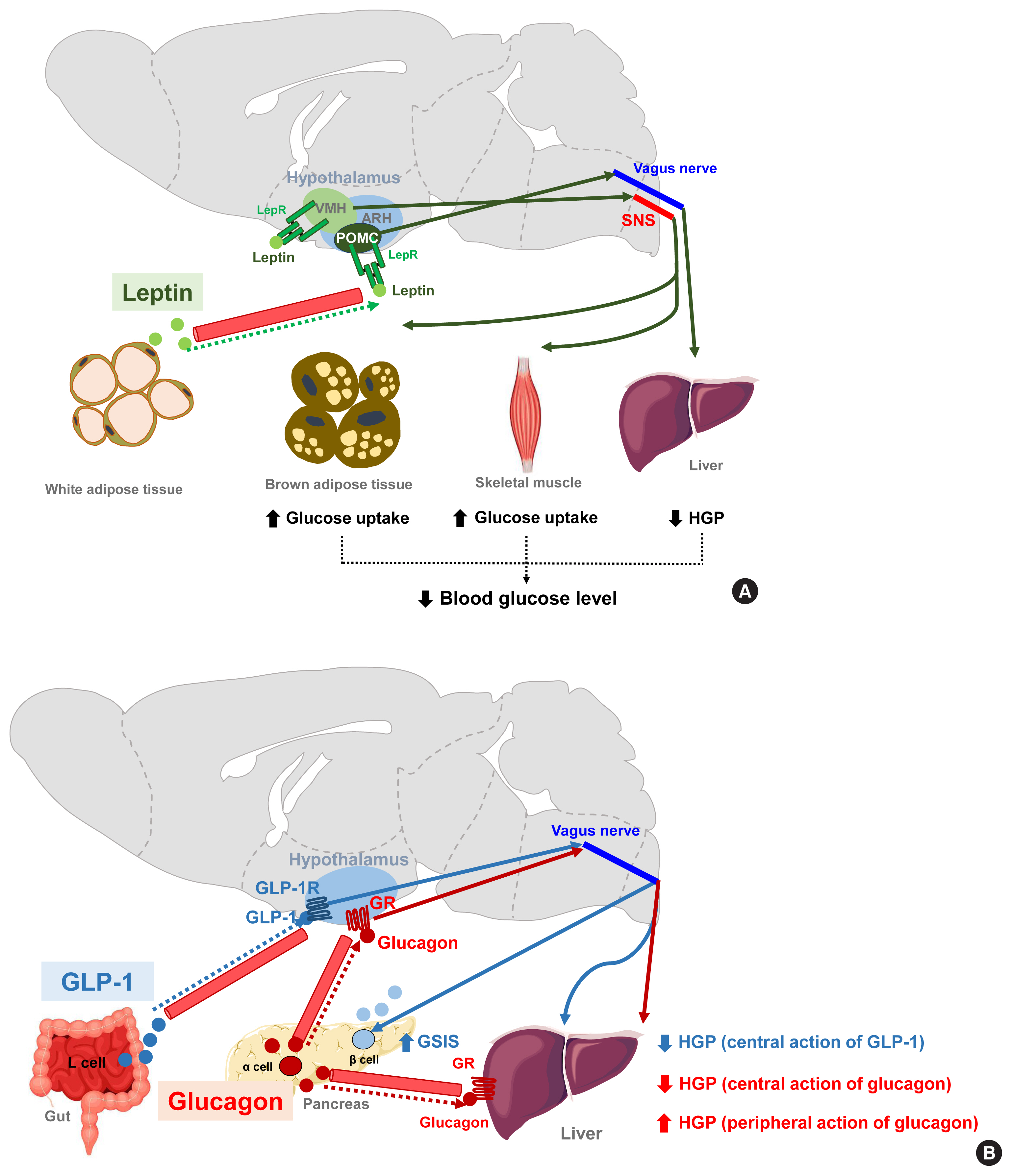
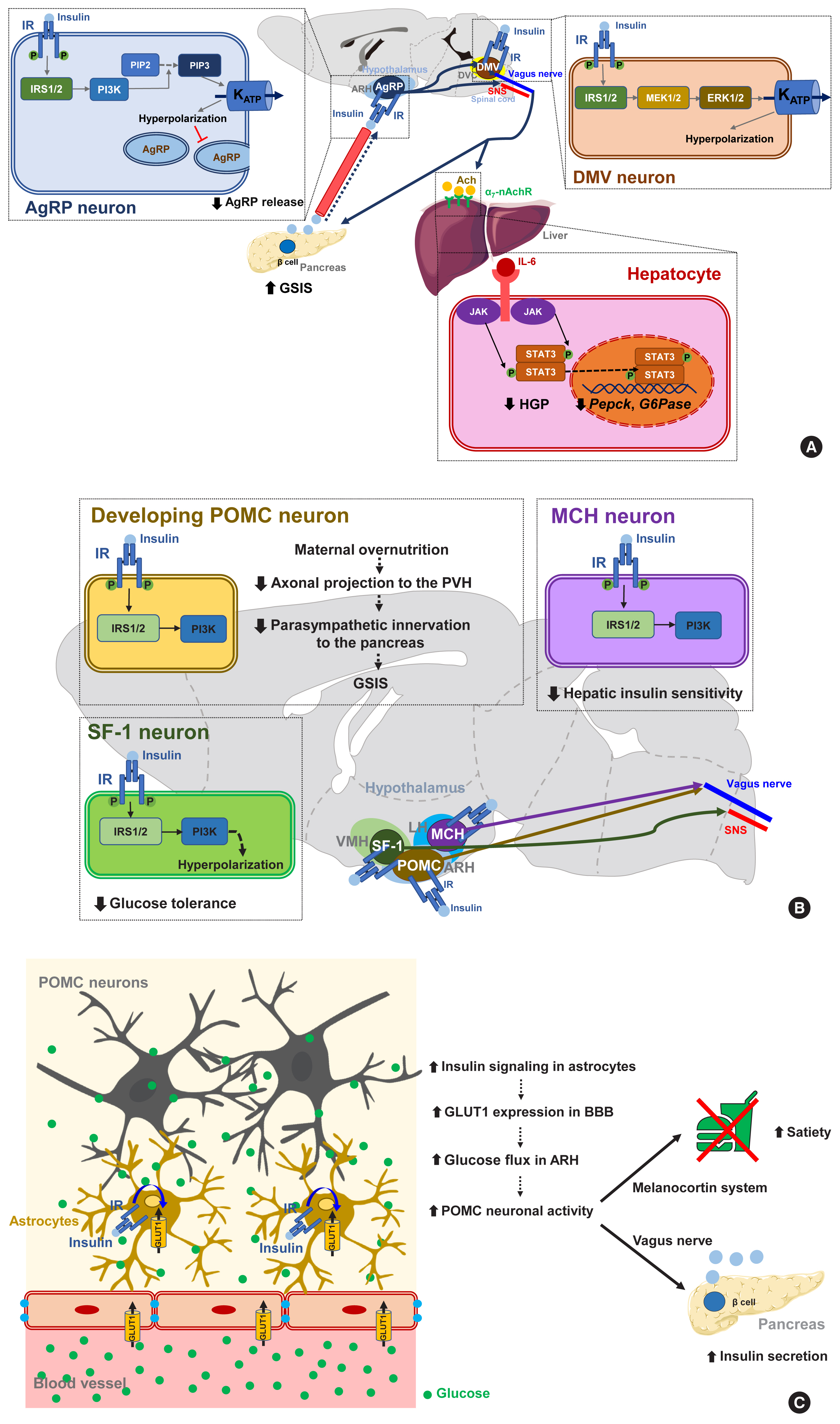
- Several lines of evidence suggest that central insulin signaling improves peripheral glucose metabolism. For example, insulin-induced suppression of HGP was reported to be attenuated when the brain IR and its downstream signaling were inhibited, regardless of the circulating insulin levels [70]. Moreover, insulin infusion into the cerebroventricle was shown to suppress HGP and this effect was transmitted to the liver via hepatic vagal innervation and the α7-nicotinic acetylcholine receptor (Fig. 3A) [71]. Thus, insulin suppresses HGP through the CNS and via vagus nerve-mediated mechanisms. ARH AgRP neurons mediate this central effect of insulin on HGP as the restoration of IR in these neurons in generalized IR null mice rescues the ability of insulin to suppress HGP [8]. In AgRP neurons, insulin inhibits neuronal activity through phosphatidyl-inositol-3 kinase (PI3K)-dependent mechanisms and via the opening of KATP channels (Fig. 3A) [72]. Other important insulin-target neurons with respect to HGP control are located in the brainstem DVC. Whereas PI3K mediates insulin signaling in the hypothalamic neurons, extracellular signal-regulated kinase (ERK) mediates insulin signaling in the DMV neurons through the direct phosphorylation and activation of KATP channels (Fig. 3A) [73]. With regard to hepatic mechanisms, central IR signaling activates hepatic signal transducer and activator of transcription 3 (STAT3) signaling through increased interleukin-6 (IL-6) production, which leads to a reduced expression of gluconeogenic genes (Pepck, G6Pase) and HGP [74]. Hence, the actions of central insulin on the HGP levels is believed to be mediated by neural KATP channel−vagus−hepatic IL-6/STAT3 signaling.
- In addition to HGP regulation, central insulin signaling may regulate systemic insulin sensitivity. In humans, intranasal insulin administration, which delivers insulin to the brain, increases peripheral fat mobilization, thermogenesis, and peripheral insulin sensitivity [75]. This insulin-mediated thermogenic regulation may underlie diet-induced thermogenesis. Consistently, the artificial activation of AgRP neurons in mice using optogenetic techniques inhibits insulin-stimulated glucose uptake in the BAT by increasing myostatin expression (Fig. 3A) [7]. Given the inhibitory effect of insulin on AgRP neuronal activity, it has been speculated that insulin may stimulate glucose uptake by the BAT through AgRP inhibition. Meanwhile, insulin inhibits lipolysis in the adipose tissue via central mechanisms and, via a process which is mediated by POMC neuronal insulin signaling, dampens the sympathetic flow to the adipose tissues [76]. As skeletal muscle is an important site of glucose processing, it is possible that central insulin signaling may regulate glucose metabolism in skeletal muscles. Indeed, a prior study has found that intracerebroventricular insulin infusion increases muscle glycogen synthesis [77], although the details of the mechanisms and neuronal circuits underlying this effect remain to be revealed.
- An additional regulatory aspect of insulin is that it acts on the brain to regulate its own secretion. Hypothalamic GK-expressing glucose-sensing neurons display multisynaptic connections to the pancreas via brainstem DMV neurons and vagal innervation [78]. The ablation of hypothalamic GLUT4-expressing neurons leads to a reduction in insulin secretion [79]. Brain insulin signaling is also required for counterregulatory responses to hypoglycemia [80]. NIRKO mice exhibit a blunted sympathoadrenal response to hypoglycemia due to reduced glucose-sensing in their hypothalamic neurons [81,82].
- It is noteworthy that insulin signaling in certain subsets of neurons can cause adverse metabolic outcomes under overnutrition conditions (Fig. 3B) [83]. Insulin signaling in POMC neurons may have a negative impact on peripheral glucose metabolism as selective IR expression in POMC neurons in whole-body IR knockout mice aggravates insulin resistance and increases HGP. Maternal overnutrition during lactation predisposes offspring to metabolism disorders in adulthood [84]. It has been further shown that maternal HFD feeding reduces the axonal projections of ARH POMC neurons to the posterior part of the PVH and also suppresses the parasympathetic innervation of pancreatic islets, leading to reduced glucose-stimulated insulin secretion (GSIS) and glucose intolerance (Fig. 3B). Interestingly, POMC-specific IR deletion in mouse pups rescues maternal overnutrition-induced disturbances in POMC neuron-to-islet circuit formation and glucose metabolism. Therefore, maternal overnutrition in the mouse may overactivate insulin signaling in POMC neurons in pups, which possibly interrupts the neural circuit organization from hypothalamic POMC neurons to pancreatic islets and causes a long-lived defect in glucose metabolism in their progeny.
- Similarly to the above results, insulin increases the electrical activity of melanin-concentrating hormone (MCH)-producing neurons in the LH, which leads to impaired glucose homeostasis through the control of hepatic insulin sensitivity and HGP (Fig. 3B) [85]. Activated insulin signaling, i.e., increased PI3K activity, is observed in the MCH neurons of HFD-fed mice [85]. Thus, insulin signaling in MCH neurons may mediate the adverse metabolic outcomes of overnutrition-associated hyperinsulinemia. On the other hand, insulin inhibits VMH SF-1 neuronal activity through a similar mechanism to AgRP neurons. Interestingly, SF-1 neuron-specific IR deletion curbs HFD-induced obesity, hyperinsulinemia, and glucose intolerance [86]. This result suggests that insulin acts on VMH SF-1 neurons to promote obesity and glucose dysregulation (Fig. 3B).
- A recent study has corroborated the importance of astrocyte insulin signaling in the brain regulation of glucose metabolism (Fig. 3C). In that report, the genetic ablation of IR in astrocytes was found to lead to impaired metabolic responses to blood glucose fluctuations [87]. Mice lacking astrocytic IRs show reduced satiety and lower insulin secretion and develop higher blood glucose levels following peritoneal glucose injection. This impairment is related to a reduced glucose flux to the hypothalamus due to lower astrocytic GLUT1 expression. Moreover, these mice have reduced hyperphagia upon exposure to hypoglycemia [87]. These results indicate that astrocyte insulin signaling critically mediates glucose-sensing and regulation by the CNS via the control of glucose transport across the BBB.
CONTRIBUTION OF CENTRAL INSULIN SIGNALING TO SYSTEMIC GLUCOSE METABOLISM
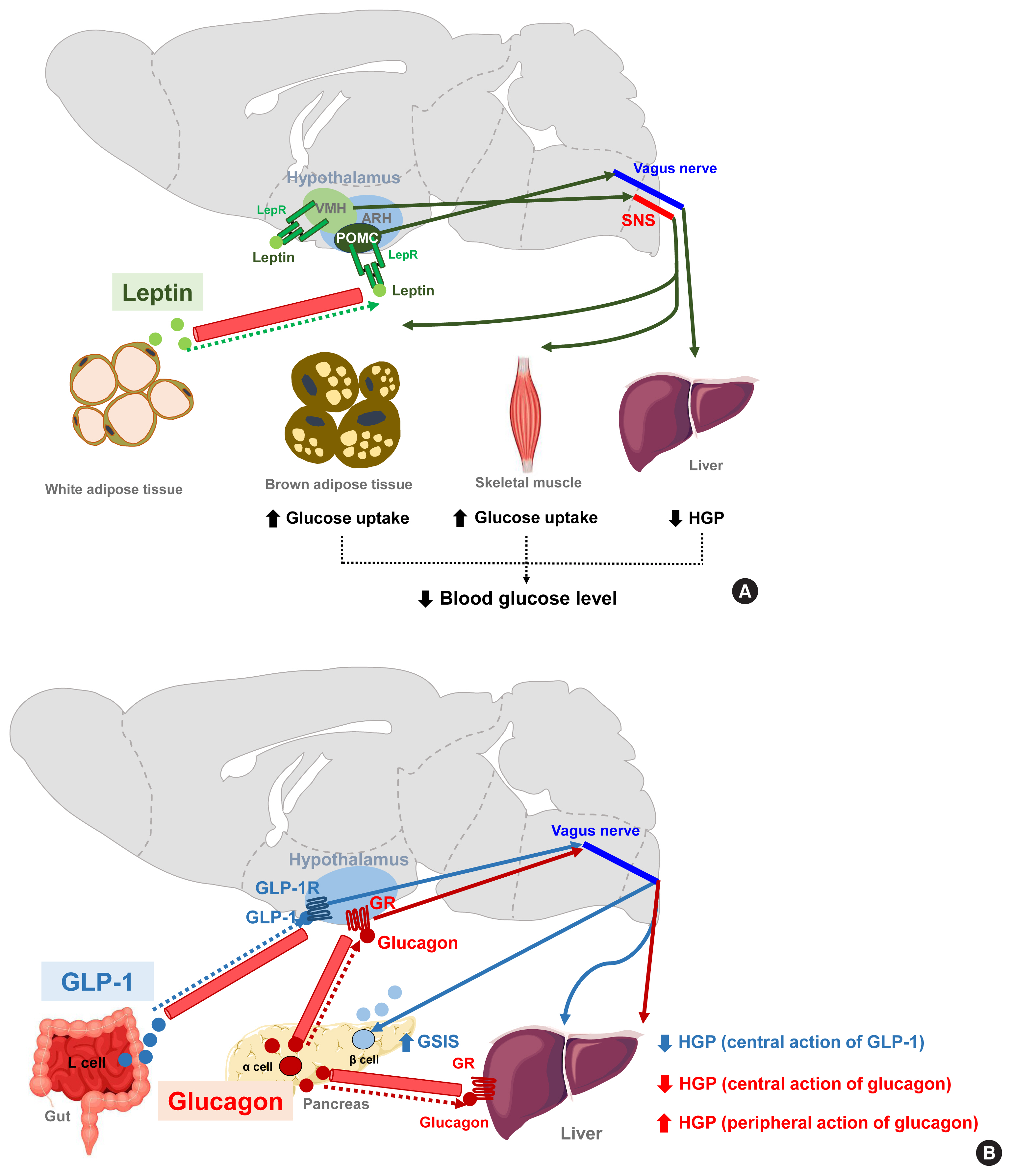
- Leptin is an adipocyte-derived hormone that regulates not only body weight but also glucose metabolism by acting on the brain. A leptin infusion of the cerebroventricle improves hyperglycemia in rats with insulin-deficient diabetes. The glucose-lowering effect of leptin is due to reduced HGP and increased insulin-independent glucose uptake into the brain, skeletal muscle, and BAT (Fig. 4A). The first-order neurons mediating leptin actions are distributed in the ARH. In leptin receptor-null mice, the reconstitution of leptin receptors, specifically in ARH POMC neurons, normalizes the blood glucose levels and these effects are independent of food intake and body weights. Moreover, mice with double knockout of insulin and leptin receptors in POMC neurons cannot suppress HGP during the hyperinsulinemic-euglycemic clamp, and these animals develop systemic insulin resistance and glucose intolerance [88]. Hence, ARH POMC neurons seem to mediate the modulation of peripheral glucose metabolism by leptin. Several lines of evidence suggest that leptin also regulates skeletal muscle glucose metabolism via the VMH neurons. Local leptin injections into the VMH increase glucose uptake in mouse skeletal muscle and improve glucose tolerance independently of the circulating insulin levels [89,90]. These effects are abolished when the sympathetic nervous system and central melanocortin receptor signaling are inhibited [16]. Central leptin injections enhance insulin-stimulated Akt phosphorylation and also activate AMPK signaling in skeletal muscle [91,92]. Both signaling events are expected to increase glucose uptake in skeletal muscle.
- GLP-1 is a gut-derived hormone with an incretin effect. In addition to its direct effects on pancreatic islets, GLP-1 augments GSIS and suppresses HGP through GLP-1 receptor signaling in the ARH (Fig. 4B) [93]. Glucagon, secreted by pancreatic α-cells, is thought to elevate blood glucose levels by acting on the hepatocytes and increasing HGP. In contrast to this peripheral action, the activation of hypothalamic glucagon receptor signaling inhibits HGP through a KATP channel-dependent mechanism (Fig. 4B) [94]. These opposing effects of central glucagon signaling on HGP may constitute a negative feedback loop that self-limits glucagon-stimulated HGP and fine-tunes the regulation of HGP by glucagon.
OTHER HORMONAL SIGNALING MECHANISMS INVOLVED IN BRAIN REGULATION OF GLUCOSE METABOLISM
- Obesity is a major risk factor for type 2 diabetes and, along with the intake of saturated fatty acids, can disrupt insulin signaling and glucose metabolism in peripheral metabolic organs. Several mechanisms such as the activation of inflammation signaling, endoplasmic reticulum stress, mitochondrial dysfunction, and accumulation of toxic lipid metabolites have been suggested to bridge obesity and metabolic disorders. Similar pathological events may also occur in the brain areas that control glucose metabolism [95].
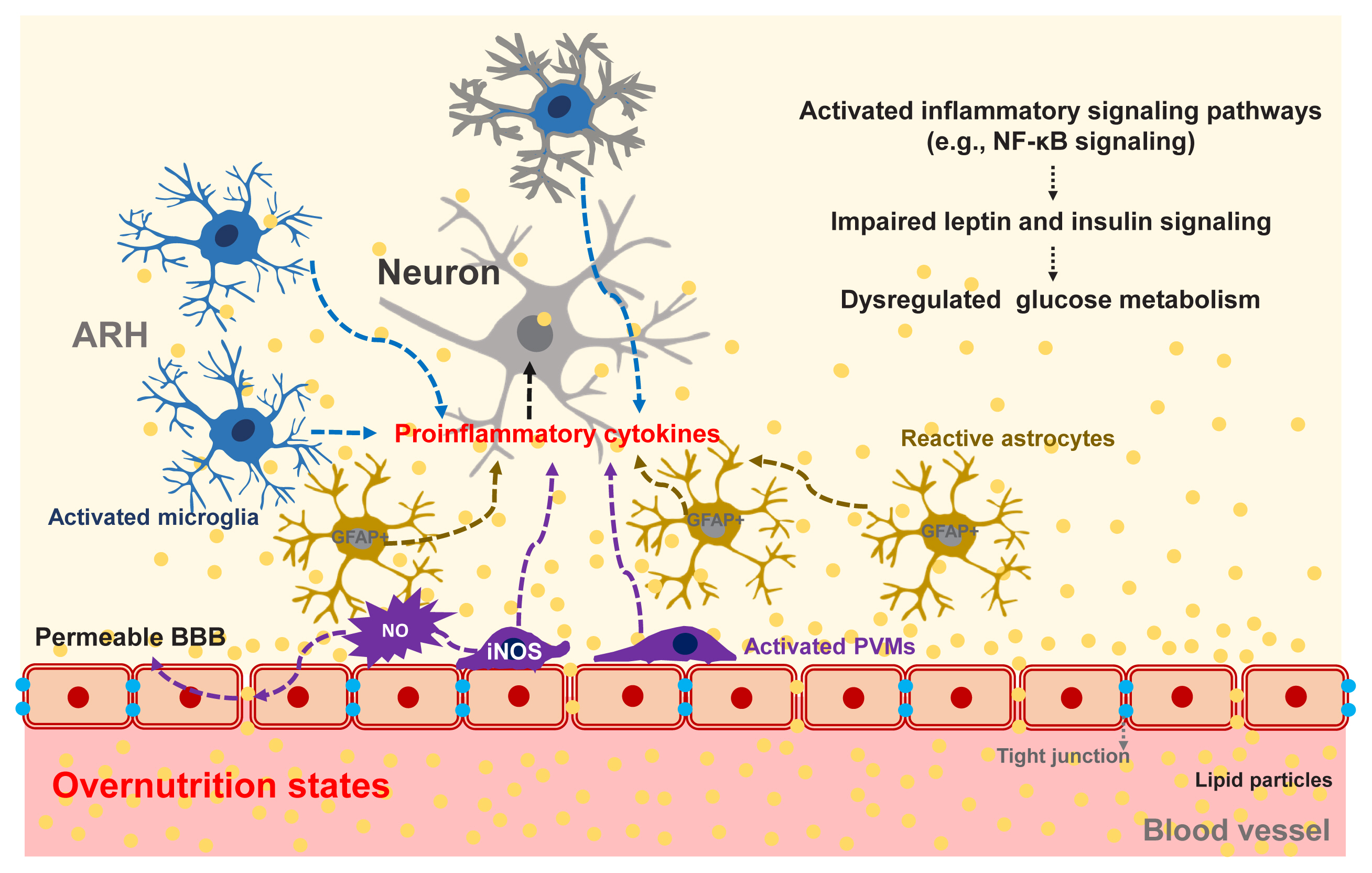
- The repeated consumption of fat-rich diets activates and accumulates microglia, which are brain-resident innate immune cells, in the ARH. Activated microglia secrete proinflammatory cytokines and elicit inflammatory signaling pathways in adjacent neurons. A considerable amount of data has now corroborated the evidence that activated inflammatory signaling hampers leptin and insulin signal transduction in hypothalamic neurons and that these signaling changes disturb the hormone-mediated hypothalamic regulation of glucose metabolism [96].
- The accumulation of GFAP-expressing reactive astrocytes, so-called astrogliosis, is another cardinal feature of obesity-associated hypothalamic changes [97]. Given the critical roles of astrocytes in hypothalamic glucose-sensing, astrogliosis may possibly contribute to glucose dysregulation in obese animals and humans. Indeed, the inhibition of nuclear factor kappa B (NF-κB) signaling in astrocytes significantly attenuates obesity progression and glucose dysmetabolism during HFD feeding [98]. However, it is unclear whether improved glucose metabolism in these mice is secondary to or independent of weight loss.
- Hypothalamic PVMs, which reside between the hypothalamic parenchyma and blood vessels, may contribute to obesity-induced alteration in glucose metabolism. As mentioned in the section on non-neuronal glucose-sensing, PVMs help to maintain glucose transport into the hypothalamus through VEGF secretion under the HFD-fed condition [83]. On the other hand, chronic HFD consumption inflames PVMs to secrete proinflammatory cytokines and boost hypothalamic inflammatory responses to fatty acids. Activated PVMs also release reactive oxygen species such as nitric oxide (NO) [99]. Enhanced NO production in PVMs may increase the permeability of BBB, leading to increased fatty acid flux in the hypothalamus [99]. As a result, fatty acids-induced hypothalamic inflammation and disturbances in hypothalamus-mediated glucose regulation may be accelerated. Supporting this notion, a blockade of inducible NO synthase (iNOS) in hypothalamic PVMs significantly improves systemic insulin resistance, the homeostatic model assessment of insulin resistance (HOMA-IR) index, and glucose intolerance in HFD-fed obese mice [99]. These findings suggest that iNOS signaling in the hypothalamic PVMs may regulate systemic glucose metabolism and insulin resistance via as yet unclear mechanisms. Collectively, overnutrition-induced glucose intolerance and insulin resistance clearly involve central mechanisms, as depicted in Fig. 5.
OBESITY-INDUCED HYPOTHALAMIC INFLAMMATION AND ITS POSSIBLE IMPACTS ON GLUCOSE HOMEOSTASIS
- This review highlights the robust evidence that the brain plays a vital role in the homeostatic regulation of glucose metabolism. The brain has highly organized machinery, differentiated by cell type and region (hypothalamic and brainstem), that can detect changes in systemic glucose metabolism. The hypothalamic and brainstem neurons also sense the peripheral metabolic state through hormones such as insulin, leptin, GLP-1, etc. In addition to neurons that have established roles in the brain, recent evidence has now revealed an essential role of the glial cells in the regulation of glucose metabolism. To achieve glucose homeostasis, the brain modulates peripheral organ glucose metabolism in many different ways, i.e., control of glucose flux in the liver and skeletal muscle, insulin/glucagon secretion in the endocrine pancreas, glucose reabsorption/secretion in the kidney, and as yet unidentified processes. The specific neurons and their circuits that are involved in each aspect of peripheral organ glucose metabolism are beginning to be revealed. A better understanding of the neuronal mechanisms of glucose homeostasis will likely lead to the discovery of novel therapeutic targets for combating hyperglycemia and hypoglycemia.
CONCLUSIONS
-
Acknowledgements
- This study was supported by grants from the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT of Korea (2020R1A2C3004843, 2020R1A4A3078962), the Asan Institute for Life Sciences (2019-IP0855-1).
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
- 1. Bernard C. Leçons de physiologie expérimentale appliquée á la médecine, faites au Collége de France; Paris: J.B. Bailliére et fils; 1855.
- 2. Mayer J. The glucostatic theory of regulation of food intake and the problem of obesity. Bull New Engl Med Cent 1952;14:43–9.PubMed
- 3. Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 1997;99:361–5.ArticlePubMedPMC
- 4. Oomura Y, Kimura K, Ooyama H, Maeno T, Iki M, Kuniyoshi M. Reciprocal activities of the ventromedial and lateral hypothalamic areas of cats. Science 1964;143:484–5.ArticlePubMed
- 5. Anand BK, Chhina GS, Sharma KN, Dua S, Singh B. Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am J Physiol 1964;207:1146–54.ArticlePubMed
- 6. Roh E, Song DK, Kim MS. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med 2016;48:e216.ArticlePubMedPMC
- 7. Steculorum SM, Ruud J, Karakasilioti I, Backes H, Engstrom Ruud L, Timper K, et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 2016;165:125–38.ArticlePubMedPMC
- 8. Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 2007;5:438–49.ArticlePubMed
- 9. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep 2012;13:1079–86.ArticlePubMedPMC
- 10. Uner AG, Kecik O, Quaresma PG, De Araujo TM, Lee H, Li W, et al. Role of POMC and AgRP neuronal activities on glycaemia in mice. Sci Rep 2019;9:13068.ArticlePubMedPMC
- 11. Chhabra KH, Adams JM, Fagel B, Lam DD, Qi N, Rubinstein M, et al. Hypothalamic POMC deficiency improves glucose tolerance despite insulin resistance by increasing glycosuria. Diabetes 2016;65:660–72.ArticlePubMed
- 12. de Souza Cordeiro LM, Elsheikh A, Devisetty N, Morgan DA, Ebert SN, Rahmouni K, et al. Hypothalamic MC4R regulates glucose homeostasis through adrenaline-mediated control of glucose reabsorption via renal GLUT2 in mice. Diabetologia 2021;64:181–94.ArticlePubMed
- 13. Shimazu T, Fukuda A, Ban T. Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature 1966;210:1178–9.ArticlePubMed
- 14. Meek TH, Nelson JT, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, et al. Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 2016;113:E2073–82.ArticlePubMedPMC
- 15. Coutinho EA, Okamoto S, Ishikawa AW, Yokota S, Wada N, Hirabayashi T, et al. Activation of SF1 neurons in the ventromedial hypothalamus by DREADD technology increases insulin sensitivity in peripheral tissues. Diabetes 2017;66:2372–86.ArticlePubMed
- 16. Toda C, Shiuchi T, Lee S, Yamato-Esaki M, Fujino Y, Suzuki A, et al. Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes 2009;58:2757–65.ArticlePubMedPMC
- 17. Gavini CK, Jones WC 2nd, Novak CM. Ventromedial hypothalamic melanocortin receptor activation: regulation of activity energy expenditure and skeletal muscle thermogenesis. J Physiol 2016;594:5285–301.ArticlePubMedPMC
- 18. Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, Diano S. UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 2016;164:872–83.ArticlePubMedPMC
- 19. Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 2013;341:1517–21.ArticlePubMedPMC
- 20. Morgan DA, McDaniel LN, Yin T, Khan M, Jiang J, Acevedo MR, et al. Regulation of glucose tolerance and sympathetic activity by MC4R signaling in the lateral hypothalamus. Diabetes 2015;64:1976–87.ArticlePubMedPMC
- 21. Shiuchi T, Haque MS, Okamoto S, Inoue T, Kageyama H, Lee S, et al. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab 2009;10:466–80.ArticlePubMed
- 22. Sanchez-Alavez M, Osborn O, Tabarean IV, Holmberg KH, Eberwine J, Kahn CR, et al. Insulin-like growth factor 1-mediated hyperthermia involves anterior hypothalamic insulin receptors. J Biol Chem 2011;286:14983–90.ArticlePubMedPMC
- 23. Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 2012;16:296–309.ArticlePubMedPMC
- 24. Ritter S, Li AJ, Wang Q, Dinh TT. Minireview: The value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 2011;152:4019–32.ArticlePubMedPMC
- 25. Schwartz GJ. The role of gastrointestinal vagal afferents in the control of food intake: current prospects. Nutrition 2000;16:866–73.ArticlePubMed
- 26. Mercer JG, Moar KM, Findlay PA, Hoggard N, Adam CL. Association of leptin receptor (OB-Rb), NPY and GLP-1 gene expression in the ovine and murine brainstem. Regul Pept 1998;75–76:271–8.Article
- 27. Georgescu T, Lyons D, Doslikova B, Garcia AP, Marston O, Burke LK, et al. Neurochemical characterization of brainstem pro-opiomelanocortin cells. Endocrinology 2020;161:bqaa032.ArticlePubMedPMC
- 28. Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, et al. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J Neurosci 2013;33:3624–32.ArticlePubMedPMC
- 29. Berglund ED, Liu T, Kong X, Sohn JW, Vong L, Deng Z, et al. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat Neurosci 2014;17:911–3.ArticlePubMedPMC
- 30. Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 2011;13:195–204.ArticlePubMedPMC
- 31. Xu H, Boychuk JA, Boychuk CR, Uteshev VV, Smith BN. Nicotine enhances inhibition of mouse vagal motor neurons by modulating excitability of premotor GABAergic neurons in the nucleus tractus solitarii. J Neurophysiol 2015;113:1165–74.ArticlePubMed
- 32. Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 2009;137:1225–34.ArticlePubMedPMC
- 33. Pozo M, Claret M. Hypothalamic control of systemic glucose homeostasis: the pancreas connection. Trends Endocrinol Metab 2018;29:581–94.ArticlePubMed
- 34. Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci 2014;8:236.ArticlePubMedPMC
- 35. Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 2002;76:403–13.PubMed
- 36. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 2007;22:241–51.ArticlePubMed
- 37. Burcelin R, Thorens B. Evidence that extrapancreatic GLUT2-dependent glucose sensors control glucagon secretion. Diabetes 2001;50:1282–9.ArticlePubMed
- 38. Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, et al. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest 2005;115:3545–53.ArticlePubMedPMC
- 39. Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci 2001;4:507–12.ArticlePubMed
- 40. Evans ML, McCrimmon RJ, Flanagan DE, Keshavarz T, Fan X, McNay EC, et al. Hypothalamic ATP-sensitive K + channels play a key role in sensing hypoglycemia and triggering counterregulatory epinephrine and glucagon responses. Diabetes 2004;53:2542–51.ArticlePubMed
- 41. McCrimmon RJ, Evans ML, Fan X, McNay EC, Chan O, Ding Y, et al. Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats. Diabetes 2005;54:3169–74.ArticlePubMed
- 42. Gyte A, Pritchard LE, Jones HB, Brennand JC, White A. Reduced expression of the KATP channel subunit, Kir6.2, is associated with decreased expression of neuropeptide Y and agouti-related protein in the hypothalami of Zucker diabetic fatty rats. J Neuroendocrinol 2007;19:941–51.ArticlePubMedPDF
- 43. Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 2006;55:412–20.ArticlePubMed
- 44. Stanley S, Domingos AI, Kelly L, Garfield A, Damanpour S, Heisler L, et al. Profiling of glucose-sensing neurons reveals that GHRH neurons are activated by hypoglycemia. Cell Metab 2013;18:596–607.ArticlePubMed
- 45. Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y. Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature 1974;247:284–6.ArticlePubMed
- 46. Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2001;50:2673–81.ArticlePubMed
- 47. Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res 1998;814:41–54.ArticlePubMed
- 48. Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurones of the rat hypothalamus. Nature 1969;222:282–4.ArticlePubMed
- 49. Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res 1984;307:109–16.ArticlePubMed
- 50. Funahashi M, Adachi A. Glucose-responsive neurons exist within the area postrema of the rat: in vitro study on the isolated slice preparation. Brain Res Bull 1993;32:531–5.ArticlePubMed
- 51. Yettefti K, Orsini JC, Perrin J. Characteristics of glycemia-sensitive neurons in the nucleus tractus solitarii: possible involvement in nutritional regulation. Physiol Behav 1997;61:93–100.ArticlePubMed
- 52. Steinbusch L, Labouebe G, Thorens B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol Metab 2015;26:455–66.ArticlePubMed
- 53. Slusser PG, Ritter RC. Increased feeding and hyperglycemia elicited by intracerebroventricular 5-thioglucose. Brain Res 1980;202:474–8.ArticlePubMed
- 54. Koekkoek LL, Mul JD, la Fleur SE. Glucose-sensing in the reward system. Front Neurosci 2017;11:716.ArticlePubMedPMC
- 55. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 1994;91:10625–9.ArticlePubMedPMC
- 56. Chari M, Yang CS, Lam CK, Lee K, Mighiu P, Kokorovic A, et al. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes 2011;60:1901–6.ArticlePubMedPMC
- 57. Goodman T, Hajihosseini MK. Hypothalamic tanycytes-masters and servants of metabolic, neuroendocrine, and neurogenic functions. Front Neurosci 2015;9:387.ArticlePubMedPMC
- 58. Rodriguez EM, Blazquez JL, Pastor FE, Pelaez B, Pena P, Peruzzo B, et al. Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol 2005;247:89–164.ArticlePubMed
- 59. Langlet F, Mullier A, Bouret SG, Prevot V, Dehouck B. Tanycyte-like cells form a blood–cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J Comp Neurol 2013;521:3389–405.ArticlePubMedPMC
- 60. Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab 2014;19:293–301.ArticlePubMedPMC
- 61. Freire-Regatillo A, Argente-Arizon P, Argente J, Garcia-Segura LM, Chowen JA. Non-neuronal cells in the hypothalamic adaptation to metabolic signals. Front Endocrinol (Lausanne) 2017;8:51.ArticlePubMedPMC
- 62. Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 2007;56:1219–27.PubMed
- 63. Sanders NM, Dunn-Meynell AA, Levin BE. Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes 2004;53:1230–6.ArticlePubMed
- 64. Elizondo-Vega R, Cortes-Campos C, Barahona MJ, Oyarce KA, Carril CA, Garcia-Robles MA. The role of tanycytes in hypothalamic glucosensing. J Cell Mol Med 2015;19:1471–82.ArticlePubMedPMC
- 65. Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 2016;165:882–95.ArticlePubMed
- 66. Winkler Z, Kuti D, Polyak A, Juhasz B, Gulyas K, Lenart N, et al. Hypoglycemia-activated hypothalamic microglia impairs glucose counterregulatory responses. Sci Rep 2019;9:6224.ArticlePubMedPMC
- 67. Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014;63:2232–43.ArticlePubMedPMC
- 68. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science 2000;289:2122–5.ArticlePubMed
- 69. Lin X, Taguchi A, Park S, Kushner JA, Li F, Li Y, et al. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J Clin Invest 2004;114:908–16.ArticlePubMedPMC
- 70. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–82.ArticlePubMed
- 71. Kimura K, Tanida M, Nagata N, Inaba Y, Watanabe H, Nagashimada M, et al. Central insulin action activates Kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep 2016;14:2362–74.ArticlePubMed
- 72. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005;434:1026–31.ArticlePubMed
- 73. Filippi BM, Yang CS, Tang C, Lam TK. Insulin activates Erk1/2 signaling in the dorsal vagal complex to inhibit glucose production. Cell Metab 2012;16:500–10.ArticlePubMed
- 74. Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 2006;3:267–75.ArticlePubMed
- 75. Benedict C, Brede S, Schioth HB, Lehnert H, Schultes B, Born J, et al. Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men. Diabetes 2011;60:114–8.ArticlePubMed
- 76. Shin AC, Filatova N, Lindtner C, Chi T, Degann S, Oberlin D, et al. Insulin receptor signaling in POMC, but not AgRP, neurons controls adipose tissue insulin action. Diabetes 2017;66:1560–71.ArticlePubMedPMC
- 77. Perrin C, Knauf C, Burcelin R. Intracerebroventricular infusion of glucose, insulin, and the adenosine monophosphate-activated kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, controls muscle glycogen synthesis. Endocrinology 2004;145:4025–33.ArticlePubMed
- 78. Chen M, Woods SC, Porte D Jr. Effect of cerebral intraventricular insulin on pancreatic insulin secretion in the dog. Diabetes 1975;24:910–4.ArticlePubMed
- 79. Ren H, Lu TY, McGraw TE, Accili D. Anorexia and impaired glucose metabolism in mice with hypothalamic ablation of Glut4 neurons. Diabetes 2015;64:405–17.ArticlePubMed
- 80. Biggers DW, Myers SR, Neal D, Stinson R, Cooper NB, Jaspan JB, et al. Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 1989;38:7–16.Article
- 81. Diggs-Andrews KA, Zhang X, Song Z, Daphna-Iken D, Routh VH, Fisher SJ. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 2010;59:2271–80.ArticlePubMedPMC
- 82. Fisher SJ, Bruning JC, Lannon S, Kahn CR. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 2005;54:1447–51.ArticlePubMed
- 83. Jais A, Bruning JC. Hypothalamic inflammation in obesity and metabolic disease. J Clin Invest 2017;127:24–32.ArticlePubMedPMC
- 84. Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 2014;156:495–509.ArticlePubMedPMC
- 85. Hausen AC, Ruud J, Jiang H, Hess S, Varbanov H, Kloppenburg P, et al. Insulin-dependent activation of MCH neurons impairs locomotor activity and insulin sensitivity in obesity. Cell Rep 2016;17:2512–21.ArticlePubMed
- 86. Klockener T, Hess S, Belgardt BF, Paeger L, Verhagen LA, Husch A, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci 2011;14:911–8.ArticlePubMedPMC
- 87. Garcia-Caceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016;166:867–80.ArticlePubMedPMC
- 88. Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab 2010;11:286–97.ArticlePubMedPMC
- 89. Shimazu T, Sudo M, Minokoshi Y, Takahashi A. Role of the hypothalamus in insulin-independent glucose uptake in peripheral tissues. Brain Res Bull 1991;27:501–4.ArticlePubMed
- 90. Sudo M, Minokoshi Y, Shimazu T. Ventromedial hypothalamic stimulation enhances peripheral glucose uptake in anesthetized rats. Am J Physiol 1991;261(3 Pt 1):E298–303.ArticlePubMed
- 91. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002;415:339–43.ArticlePubMed
- 92. Shiuchi T, Toda C, Okamoto S, Coutinho EA, Saito K, Miura S, et al. Induction of glucose uptake in skeletal muscle by central leptin is mediated by muscle β2-adrenergic receptor but not by AMPK. Sci Rep 2017;7:15141.ArticlePubMedPMC
- 93. Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008;57:2046–54.ArticlePubMedPMC
- 94. Mighiu PI, Yue JT, Filippi BM, Abraham MA, Chari M, Lam CK, et al. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med 2013;19:766–72.ArticlePubMed
- 95. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 2017;127:1–4.ArticlePubMedPMC
- 96. Lee CH, Suk K, Yu R, Kim MS. Cellular contributors to hypothalamic inflammation in obesity. Mol Cells 2020;43:431–7.PubMedPMC
- 97. Gonzalez-Garcia I, Garcia-Caceres C. Hypothalamic astrocytes as a specialized and responsive cell population in obesity. Int J Mol Sci 2021;22:6176.ArticlePubMedPMC
- 98. Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab 2017;6:366–73.ArticlePubMedPMC
- 99. Lee CH, Kim HJ, Lee YS, Kang GM, Lim HS, Lee SH, et al. Hypothalamic macrophage inducible nitric oxide synthase mediates obesity-associated hypothalamic inflammation. Cell Rep 2018;25:934–46.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Sympathetic nerve-enteroendocrine L cell communication modulates GLP-1 release, brain glucose utilization, and cognitive function
Wenran Ren, Jianhui Chen, Wenjing Wang, Qingqing Li, Xia Yin, Guanglei Zhuang, Hong Zhou, Wenwen Zeng
Neuron.2024; 112(6): 972. CrossRef - Hypothalamic astrocyte NAD+ salvage pathway mediates the coupling of dietary fat overconsumption in a mouse model of obesity
Jae Woo Park, Se Eun Park, Wuhyun Koh, Won Hee Jang, Jong Han Choi, Eun Roh, Gil Myoung Kang, Seong Jun Kim, Hyo Sun Lim, Chae Beom Park, So Yeon Jeong, Sang Yun Moon, Chan Hee Lee, Sang Yeob Kim, Hyung Jin Choi, Se Hee Min, C. Justin Lee, Min-Seon Kim
Nature Communications.2024;[Epub] CrossRef - Efficacy of metformin and electrical pulses in breast cancer MDA-MB-231 cells
Praveen Sahu, Ignacio G. Camarillo, Raji Sundararajan
Exploration of Targeted Anti-tumor Therapy.2024; 5(1): 54. CrossRef - Redox imbalance and metabolic defects in the context of Alzheimer disease
Fabio Di Domenico, Chiara Lanzillotta, Marzia Perluigi
FEBS Letters.2024;[Epub] CrossRef - Obesity-associated microglial inflammatory activation paradoxically improves glucose tolerance
John D. Douglass, Kelly M. Ness, Martin Valdearcos, Alice Wyse-Jackson, Mauricio D. Dorfman, Jeremy M. Frey, Rachael D. Fasnacht, Olivia D. Santiago, Anzela Niraula, Jineta Banerjee, Megan Robblee, Suneil K. Koliwad, Joshua P. Thaler
Cell Metabolism.2023; 35(9): 1613. CrossRef - Phenotypic screening using waveform analysis of synchronized calcium oscillations in primary cortical cultures
Richi Sakaguchi, Saki Nakamura, Hiroyuki Iha, Masaki Tanaka, Ming Tatt Lee
PLOS ONE.2023; 18(4): e0271782. CrossRef - Effects of Escitalopram on the Functional Neural Circuits in an Animal Model of Adolescent Depression
Se Jong Oh, Namhun Lee, Kyung Rok Nam, Kyung Jun Kang, Sang Jin Han, Jae Yong Choi
Molecular Imaging and Biology.2023; 25(4): 735. CrossRef - A Survey of Deep Learning for Alzheimer’s Disease
Qinghua Zhou, Jiaji Wang, Xiang Yu, Shuihua Wang, Yudong Zhang
Machine Learning and Knowledge Extraction.2023; 5(2): 611. CrossRef - Potassium channels in behavioral brain disorders. Molecular mechanisms and therapeutic potential: A narrative review
Kazi Asraful Alam, Pernille Svalastoga, Aurora Martinez, Jeffrey Colm Glennon, Jan Haavik
Neuroscience & Biobehavioral Reviews.2023; 152: 105301. CrossRef - Knockout of Nur77 Leads to Amino Acid, Lipid, and Glucose Metabolism Disorders in Zebrafish
Yang Xu, Juanjuan Tian, Qi Kang, Hang Yuan, Chengdong Liu, Zhehui Li, Jie Liu, Mingyu Li
Frontiers in Endocrinology.2022;[Epub] CrossRef - Effects of the POMC System on Glucose Homeostasis and Potential Therapeutic Targets for Obesity and Diabetes
Dan Yang, Xintong Hou, Guimei Yang, Mengnan Li, Jian Zhang, Minmin Han, Yi Zhang, Yunfeng Liu
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy.2022; Volume 15: 2939. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite