
Search
- Page Path
- HOME > Search
Original Article
- Adrenal Gland
- Clinical and Molecular Characteristics of PRKACA L206R Mutant Cortisol-Producing Adenomas in Korean Patients
- Insoon Jang, Su-jin Kim, Ra-Young Song, Kwangsoo Kim, Seongmin Choi, Jang-Seok Lee, Min-Kyeong Gwon, Moon Woo Seong, Kyu Eun Lee, Jung Hee Kim
- Endocrinol Metab. 2021;36(6):1287-1297. Published online December 2, 2021
- DOI: https://doi.org/10.3803/EnM.2021.1217
- 3,890 View
- 127 Download
-
 Abstract
Abstract
 PDF
PDF Supplementary Material
Supplementary Material PubReader
PubReader  ePub
ePub - Background
An activating mutation (c.617A>C/p.Lys206Arg, L206R) in protein kinase cAMP-activated catalytic subunit alpha (PRKACA) has been reported in 35% to 65% of cases of cortisol-producing adenomas (CPAs). We aimed to compare the clinical characteristics and transcriptome analysis between PRKACA L206R mutants and wild-type CPAs in Korea.
Methods
We included 57 subjects with CPAs who underwent adrenalectomy at Seoul National University Hospital. Sanger sequencing for PRKACA was conducted in 57 CPA tumor tissues. RNA sequencing was performed in 13 fresh-frozen tumor tissues.
Results
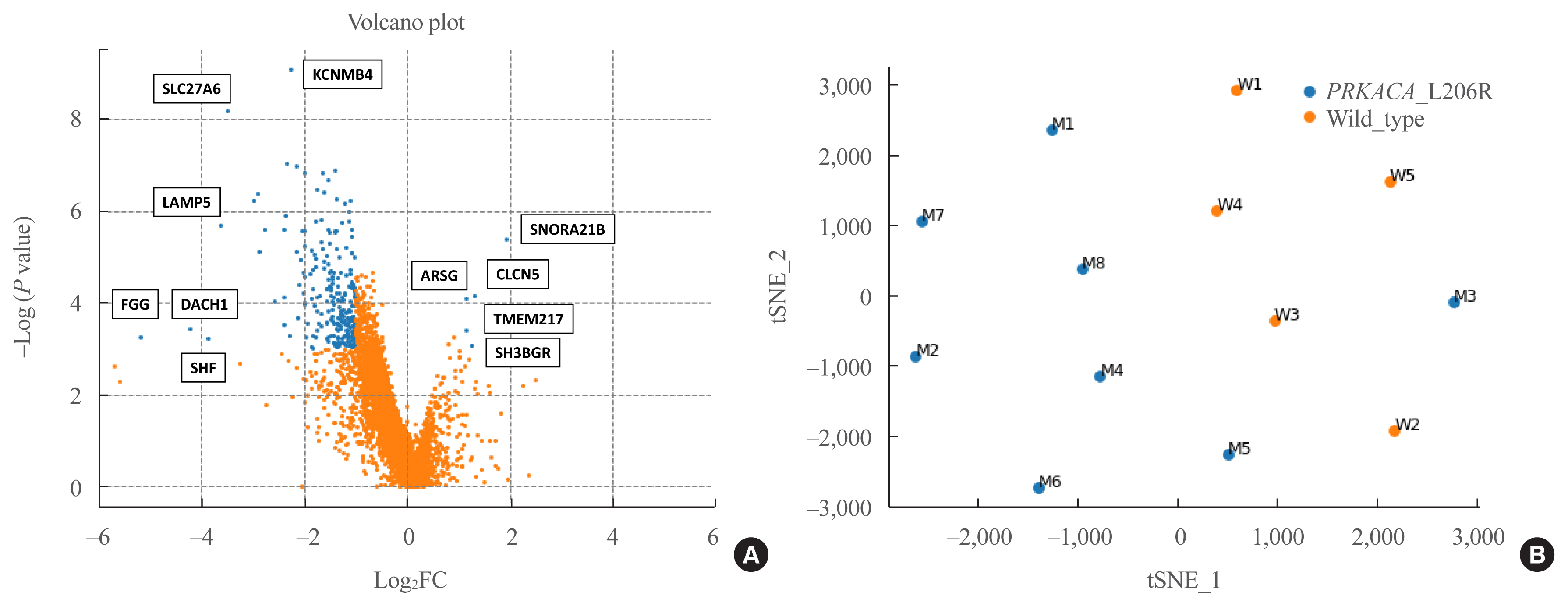
The prevalence of the PRKACA L206R mutation was 51% (29/57). The mean age of the study subjects was 42±12 years, and 87.7% (50/57) of the patients were female. Subjects with PRKACA L206R mutant CPAs showed smaller adenoma size (3.3±0.7 cm vs. 3.8±1.2 cm, P=0.059) and lower dehydroepiandrosterone sulfate levels (218±180 ng/mL vs. 1,511±3,307 ng/mL, P=0.001) than those with PRKACA wild-type CPAs. Transcriptome profiling identified 244 differentially expressed genes (DEGs) between PRKACA L206R mutant (n=8) and wild-type CPAs (n=5), including five upregulated and 239 downregulated genes in PRKACA L206R mutant CPAs (|fold change| ≥2, P<0.05). Among the upstream regulators of DEGs, CTNNB1 was the most significant transcription regulator. In several pathway analyses, the Wnt signaling pathway was downregulated and the steroid biosynthesis pathway was upregulated in PRKACA mutants. Protein-protein interaction analysis also showed that PRKACA downregulates Wnt signaling and upregulates steroid biosynthesis.
Conclusion
The PRKACA L206R mutation in CPAs causes high hormonal activity with a limited proliferative capacity, as supported by transcriptome profiling.

Case Report
- A Case of Adrenocortical Adenoma Associated with Incidental Pheochromocytoma.
- Sung Jun Hong, Young Sik Choi, Yo Han Park, Byung Cheol Yoon, Young Hwan Bae, Seon Ja Park, Ja Young Koo
- J Korean Endocr Soc. 1996;11(4):531-537. Published online November 7, 2019
- 1,183 View
- 44 Download
-
Abstract
PDF
- The coexistence of adrenal cortical tumor and pheochromocytoma was extremely rare. A total of 25 reported cases showing evidence of hyperfuction of the adrenal cortex and pheochromocytoma were noted in the literature. Of those twenty cases were coexistence of pheochromocytoma and adrenocortical hyperplasia and only five cases were coincident pheochromocytoma and adreno-cortical adenoma. Recently, we experienced a case of adrenocortical adenoma associated with incidental pheochrmocytoma. A 55-year-old woman complained of progressive weight gain and epigastric discomfort. Hormonal and radiologic studies revealed Cushings syndrome with a left adrenal tumor. Adrenalectomy was performed and the gland actually had two nodules on its surface, one pheochromocytoma and the other cortical adenoma. This patient was the first case of pheochromocytoma with adrenocortical adenoma in Korea. We report the case with a review of literature.
Letter
- Adrenal gland
- An Ectopic Cortisol-Producing Adrenocortical Adenoma Masquerading as a Liposarcoma in the Pararenal Space
- Sunyoung Kang, Seung Shin Park, Jae Hyun Bae, Kyu Eun Lee, Jung Hee Kim, Chan Soo Shin
- Endocrinol Metab. 2018;33(3):423-424. Published online August 14, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.3.423
- 2,835 View
- 43 Download
Case Reports
- Adrenal gland
- Bilateral Adrenocortical Masses Producing Aldosterone and Cortisol Independently
- Seung-Eun Lee, Jae Hyeon Kim, You-Bin Lee, Hyeri Seok, In Seub Shin, Yeong Hee Eun, Jung-Han Kim, Young Lyun Oh
- Endocrinol Metab. 2015;30(4):607-613. Published online December 31, 2015
- DOI: https://doi.org/10.3803/EnM.2015.30.4.607
- 4,114 View
- 46 Download
- 5 Web of Science
- 4 Crossref
-
Abstract
PDFPubReader
A 31-year-old woman was referred to our hospital with symptoms of hypertension and bilateral adrenocortical masses with no feature of Cushing syndrome. The serum aldosterone/renin ratio was elevated and the saline loading test showed no suppression of the plasma aldosterone level, consistent with a diagnosis of primary hyperaldosteronism. Overnight and low-dose dexamethasone suppression tests showed no suppression of serum cortisol, indicating a secondary diagnosis of subclinical Cushing syndrome. Adrenal vein sampling during the low-dose dexamethasone suppression test demonstrated excess secretion of cortisol from the left adrenal mass. A partial right adrenalectomy was performed, resulting in normalization of blood pressure, hypokalemia, and high aldosterone level, implying that the right adrenal mass was the main cause of the hyperaldosteronism. A total adrenalectomy for the left adrenal mass was later performed, resulting in a normalization of cortisol level. The final diagnosis was bilateral adrenocortical adenomas, which were secreting aldosterone and cortisol independently. This case is the first report of a concurrent cortisol-producing left adrenal adenoma and an aldosterone-producing right adrenal adenoma in Korea, as demonstrated by adrenal vein sampling and sequential removal of adrenal masses.
-
Citations
Citations to this article as recorded by
- Different cell compositions and a novel somatic KCNJ5 variant found in a patient with bilateral adrenocortical adenomas secreting aldosterone and cortisol
Liling Zhao, Jinjing Wan, Yujun Wang, Wenjun Yang, Qi Liang, Jinrong Wang, Ping Jin
Frontiers in Endocrinology.2023;[Epub] CrossRef - Adrenal Vein Cortisol to Metanephrine Ratio for Localizing ACTH-Independent Cortisol-Producing Adenoma: A Case Report
Rishi Raj, Philip A Kern, Neelima Ghanta, Edilfavia M Uy, Kamyar Asadipooya
Journal of the Endocrine Society.2021;[Epub] CrossRef - Adrenal Venous Sampling for Subtype Diagnosis of Primary Hyperaldosteronism
Mitsuhide Naruse, Akiyo Tanabe, Koichi Yamamoto, Hiromi Rakugi, Mitsuhiro Kometani, Takashi Yoneda, Hiroki Kobayashi, Masanori Abe, Youichi Ohno, Nobuya Inagaki, Shoichiro Izawa, Masakatsu Sone
Endocrinology and Metabolism.2021; 36(5): 965. CrossRef - Hypercortisolism and primary aldosteronism caused by bilateral adrenocortical adenomas: a case report
Kaiyun Ren, Jia Wei, Qilin Liu, Yuchun Zhu, Nianwei Wu, Ying Tang, Qianrui Li, Qianying Zhang, Yerong Yu, Zhenmei An, Jing Chen, Jianwei Li
BMC Endocrine Disorders.2019;[Epub] CrossRef
- Different cell compositions and a novel somatic KCNJ5 variant found in a patient with bilateral adrenocortical adenomas secreting aldosterone and cortisol
- Adrenal gland
- Untreated Congenital Adrenal Hyperplasia with 17-α Hydroxylase/17,20-Lyase Deficiency Presenting as Massive Adrenocortical Tumor
- Su Jin Lee, Je Eun Song, Sena Hwang, Ji-Yeon Lee, Hye-Sun Park, Seunghee Han, Yumie Rhee
- Endocrinol Metab. 2015;30(3):408-413. Published online August 4, 2015
- DOI: https://doi.org/10.3803/EnM.2015.30.3.408
- 4,223 View
- 47 Download
- 3 Web of Science
- 4 Crossref
-
Abstract
PDFPubReader
Congenital adrenal hyperplasia (CAH) with 17α-hydroxylase/17,20-lyase deficiency is usually characterized by hypertension and primary amenorrhea, sexual infantilism in women, and pseudohermaphroditism in men. hypertension, and sexual infantilism in women and pseudohermaphroditism in men. In rare cases, a huge adrenal gland tumor can present as a clinical manifestation in untreated CAH. Adrenal cortical adenoma is an even more rare phenotype in CAH with 17α-hydroxylase/17,20-lyase deficiency. A 36-year-old female presented with hypertension and abdominal pain caused by a huge adrenal mass. Due to mass size and symptoms, left adrenalectomy was performed. After adrenalectomy, blood pressure remained high. Based on hormonal and genetic evaluation, the patient was diagnosed as CAH with 17α-hydroxylase/17,20-lyase deficiency. The possibility of a tumorous change in the adrenal gland due to untreated CAH should be considered. It is important that untreated CAH not be misdiagnosed as primary adrenal tumor as these conditions require different treatments. Adequate suppression of adrenocorticotropic hormone (ACTH) in CAH is also important to treat and to prevent the tumorous changes in the adrenal gland. Herein, we report a case of untreated CAH with 17α-hydroxylase/17,20-lyase deficiency presenting with large adrenal cortical adenoma and discuss the progression of adrenal gland hyperplasia due to inappropriate suppression of ACTH secretion.
-
Citations
Citations to this article as recorded by- Congenital adrenal hyperplasia disorder due to 17 α-hydroxylase deficiency: a case report
Yunling Tian, Lijie Hou, Shulan Xiang, Xuguang Tian, Jinhui Xu
Gynecological Endocrinology.2023;[Epub] CrossRef - Landscape of Adrenal Tumours in Patients with Congenital Adrenal Hyperplasia
Mara Carsote, Ana-Maria Gheorghe, Claudiu Nistor, Alexandra-Ioana Trandafir, Oana-Claudia Sima, Anca-Pati Cucu, Adrian Ciuche, Eugenia Petrova, Adina Ghemigian
Biomedicines.2023; 11(11): 3081. CrossRef - 17α-Hydroxylase/17,20-Lyase Deficiency in 46,XY: Our Experience and Review of Literature
Madhur Maheshwari, Sneha Arya, Anurag Ranjan Lila, Vijaya Sarathi, Rohit Barnabas, Khushnandan Rai, Vishwambhar Vishnu Bhandare, Saba Samad Memon, Manjiri Pramod Karlekar, Virendra Patil, Nalini S Shah, Ambarish Kunwar, Tushar Bandgar
Journal of the Endocrine Society.2022;[Epub] CrossRef - 17α-hydroxylase Deficiency Mimicking Hyperaldosteronism by Aldosterone-producing Adrenal Adenoma
Yun Kyung Cho, Hyeseon Oh, Sun-myoung Kang, Sujong An, Jin-Young Huh, Ji-Hyang Lee, Woo Je Lee
The Korean Journal of Medicine.2016; 91(2): 191. CrossRef
- Congenital adrenal hyperplasia disorder due to 17 α-hydroxylase deficiency: a case report
- Two Case of Primary Aldosteronism Induced by Aldosterone Producing Adrenal Adenoma in a Family.
- Young Rock Jang, Sei Hyun Kim, Young Sil Eom, Ki Young Lee
- Endocrinol Metab. 2012;27(4):329-333. Published online December 20, 2012
- DOI: https://doi.org/10.3803/EnM.2012.27.4.329
- 1,647 View
- 23 Download
-
Abstract
PDF
- Primary aldosteronism, is defined as a group of disorders characterized by the excess of aldosteron, with suppressed rennin activity, resulting in hypertension and hypokalemia. In most cases, primary aldosteronism is sporadic due to a unilateral adrenal adenoma or bilateral adrenal hyperplasia. Familial hyperaldosteronism is a rare cause of primary aldosteronism and its prevalence has not been established well. We describe two cases of primary aldosteronism in a family involving a sister and brother due to an aldosterone producing adenoma in the left adrenal gland. Their hypokalemia and hypertension were cured by complete resection of the adrenal adenoma. Genetic analyses could not be done because of patients' rejection.
- A Case of Adrenal Cystic Pheochromocytoma with Contralateral Adrenocortical Adenoma Causing Subclinical Cushing's Syndrome.
- Chang Jun Park, Joo Wan Seo, Hyeog Gyu Seoung, Jung Hee Koh, Yong Jae Lee, Bo Hyun Kim, In Ju Kim
- Endocrinol Metab. 2012;27(4):323-328. Published online December 20, 2012
- DOI: https://doi.org/10.3803/EnM.2012.27.4.323
- 1,957 View
- 25 Download
- 1 Crossref
-
Abstract
PDF
- Bilateral adrenal neoplasms are associated with metastatic cancer, pheochromocytoma and lymphoma. The coexistence of a unilateral functioning adrenocortical adenoma with contralateral pheochromocytoma is extremely rare. A 52-year-old woman complained of fatigue, headache, palpitation, and progressive weight gain. Hormonal assessment demonstrated high 24 hours urine epinephrine, norepinephrine, and free cortisol. A dexamethasone suppression test (overnight 1 mg, low dose 2 mg) showed insuppressible cortisol. Computerized tomographic scanning revealed a bilateral adrenal tumor. To preserve adrenal function, right adrenalectomy along with left adrenal tumorectomy was performed. Histological finding of the right adrenal tumor was pheochromocytoma and the left adrenal tumor was adrenocortical adenoma. This patient was the first case of a functional adrenocortical adenoma with contralateral cystic pheochromocytoma in Korea. We report the case with a review of the literature.
-
Citations
Citations to this article as recorded by- A Case of Bilateral ACTH-independent Adrenal Adenomas with Cushing's Syndrome Treated by Ipsilateral Total and Contralateral Partial Laparoscopic Adrenalectomy
Seung Ah Park, Dong min Jung, Soon young Kim, Nan Young Choi, Tae-jun Kim, Yong kyun Kim, Seong kyun Na, Chul Sik Kim, Seong Jin Lee, Sung-Hee Ihm, Jun Goo Kang
The Korean Journal of Obesity.2013; 22(4): 254. CrossRef
- A Case of Bilateral ACTH-independent Adrenal Adenomas with Cushing's Syndrome Treated by Ipsilateral Total and Contralateral Partial Laparoscopic Adrenalectomy
Original Article
- Characterization of Incidentally Detected Adrenal Pheochromocytoma.
- Ye An Kim, Yul Hwangbo, Min Joo Kim, Hyung Jin Choi, Je Hyun Seo, Yenna Lee, Soo Heun Kwak, Eu Jeong Ku, Tae Jung Oh, Eun Roh, Jae Hyun Bae, Jung Hee Kim, Kyoung Soo Park, Seong Yeon Kim
- Endocrinol Metab. 2012;27(2):132-137. Published online June 20, 2012
- DOI: https://doi.org/10.3803/EnM.2012.27.2.132
- 2,286 View
- 28 Download
- 3 Crossref
-
Abstract
PDF
- BACKGROUND
In approach to an adrenal incidentaloma, early exclusion of pheochromocytoma is clinically important, due to the risk of catecholamine crisis. The aims of this study are to investigate the characteristics of incidentally detected pheochromocytomas, compared with that of the other adrenal incidentalomas, and to compare these characteristics with those of symptomatic pheochromocytomas. METHODS: In this retrospective study, we reviewed the medical records of 198 patients with adrenal incidentaloma from 2001 to 2010. We analyzed the clinical, laboratory and radiological data of pheochromocytomas, in comparison with those of the other adrenal incidentalomas. We also compared the characteristics of these incidentally detected pheochromocytomas with the medical records of 28 pathologically proven pheochromocytomas, diagnosed based on typical symptoms. RESULTS: Among the 198 patients with adrenal incidentaloma, nineteen patients were diagnosed with pheochromocytoma. Pheochromocytomas showed larger size and higher Hounsfield unit at precontrast computed tomography (CT) than did non-pheochromocytomas. All pheochromocytomas were larger than 2.0 cm, and the Hounsfield units were 19 or higher in precontrast CT. When both criteria of size > 2.0 cm and Hounsfield unit > 19 were met, the sensitivity and specificity for the diagnosis of pheochromocytoma were 100% and 79.3%, respectively. Compared with patients with pheochromocytoma, diagnosed based on typical symptoms, patients with incidentally detected pheochromocytoma were older, presented less often with hypertension, and showed lower levels of 24-hour urine metanephrine. CONCLUSION: Adrenal incidentaloma with < 2.0 cm in size or < or = 19 Hounsfield units in precontrast CT imaging was less likely to be a pheochromocytoma. Patients with incidentally discovered pheochromocytoma showed lower catecholamine metabolites, compared with those patients with symptomatic pheochromocytoma. -
Citations
Citations to this article as recorded by- Guidelines for the Management of Adrenal Incidentaloma: the Korean Endocrine Society, Committee of Clinical Practice Guidelines
Jung-Min Lee, Mee Kyoung Kim, Seung-Hyun Ko, Jung-Min Koh, Bo-Yeon Kim, Sang-Wan Kim, Soo-Kyung Kim, Hae-Jin Kim, Ohk-Hyun Ryu, Juri Park, Jung-Soo Lim, Seong Yeon Kim, Young Kee Shong, Soon Jib Yoo
The Korean Journal of Medicine.2017; 92(1): 4. CrossRef - Clinical Guidelines for the Management of Adrenal Incidentaloma
Jung-Min Lee, Mee Kyoung Kim, Seung-Hyun Ko, Jung-Min Koh, Bo-Yeon Kim, Sang Wan Kim, Soo-Kyung Kim, Hae Jin Kim, Ohk-Hyun Ryu, Juri Park, Jung Soo Lim, Seong Yeon Kim, Young Kee Shong, Soon Jib Yoo
Endocrinology and Metabolism.2017; 32(2): 200. CrossRef - Characterization of Incidentally Detected Adrenal Pheochromocytoma
Soon Jib Yoo, Woohyeon Kim
Endocrinology and Metabolism.2012; 27(2): 116. CrossRef
- Guidelines for the Management of Adrenal Incidentaloma: the Korean Endocrine Society, Committee of Clinical Practice Guidelines
Case Reports
- A Case of Adrenocortical Adenoma Causing Subclinical Cushing's Syndrome Mistaken for Liddle's Syndrome.
- Kyu Hong Kim, Kwang Hyun Kim, Ho Yoel Ryu, Soo Min Nam, Mi Young Lee, Jang Hyun Koh, Jang Yel Shin, Soon Hee Jung, Choon Hee Chung
- J Korean Endocr Soc. 2006;21(1):58-62. Published online February 1, 2006
- DOI: https://doi.org/10.3803/jkes.2006.21.1.58
- 2,082 View
- 25 Download
- 1 Crossref
-
Abstract
PDF
- Subclinical Cushing's syndrome is defined as an autonomous cortisol hyperproduction without specific clinical signs of cortisol excess, but detectable biochemically as derangements of the hypothalamic-pituitary-adrenal axis function. We report a case of a 33-year-old woman with subclinical Cushing's syndrome caused by left adrenocortical adenoma, mistaken for Liddle's syndrome. The patient complained of fatigue. Laboratory findings showed metabolic alkalosis, hypokalemia, high TTKG (transtubular K concentration gradient), low plasma renin activity, and low serum aldosterone level, that findings implied as Liddle's syndrome. So we performed further study. Hormonal and radiologic studies revealed subclinical Cushing's syndrome with a left adrenal mass. The adrenal mass was resected and pathologically diagnosed as adrenocortical adenoma. After the resection of the left adrenal mass, patient's hormonal levels showed normal range.
-
Citations
Citations to this article as recorded by- Missed Ipsilateral Adrenal Adenoma With Recurrent Hypercortisolism After Prior Left Adrenalectomy
Jihoon Kim, Hae Kyung Kim, Choon Hee Chung
Journal of Korean Medical Science.2022;[Epub] CrossRef
- Missed Ipsilateral Adrenal Adenoma With Recurrent Hypercortisolism After Prior Left Adrenalectomy
- A Case of Black Adrenocortical Adenoma Causing Cushing's Syndrome with Contralateral Nonfuncioning Adenoma.
- Do Joon Park, Kyung Soo Park, Kyung Jae Nam, Sung Yeon Kim, Bo Yeon Cho, Hong Gyu Lee, Yeo Kyu Yoon, Seung Keun Oh
- J Korean Endocr Soc. 1999;14(2):410-417. Published online January 1, 2001
- 1,023 View
- 17 Download
-
Abstract
PDF
- We report herein the case of a 38-year-old woman with Cushings syndrome caused by bilateral adrenocortical adenomas. The adrenal tumor on the left side hypersecreted cortisol and no findings of cortisol hypersecretion from the adrenal tumor on the right side were observed on bilateral adrenal vein samplings. Both adrenal tumors were resected and histologically without any findings of nodular hyperplasia. The left adrenal tumor was histologically diagnosed as a so-called black adenoma. These data imply that the adrenal adenomas developed primarily from the adrenal gland itself, and that one of the tumors was well differentiated and secreted excess hormones, while the other remained in cell proliferation without hypersecretion.
 First
First Prev
Prev


