Regulation of Osteoblast Metabolism by Wnt Signaling
Article information

Abstract
Wnt/β-catenin signaling plays a critical role in the achievement of peak bone mass, affecting the commitment of mesenchymal progenitors to the osteoblast lineage and the anabolic capacity of osteoblasts depositing bone matrix. Recent studies suggest that this evolutionarily-conserved, developmental pathway exerts its anabolic effects in part by coordinating osteoblast activity with intermediary metabolism. These findings are compatible with the cloning of the gene encoding the low-density lipoprotein related receptor-5 (LRP5) Wnt co-receptor from a diabetes-susceptibility locus and the now well-established linkage between Wnt signaling and metabolism. In this article, we provide an overview of the role of Wnt signaling in whole-body metabolism and review the literature regarding the impact of Wnt signaling on the osteoblast's utilization of three different energy sources: fatty acids, glucose, and glutamine. Special attention is devoted to the net effect of nutrient utilization and the mode of regulation by Wnt signaling. Mechanistic studies indicate that the utilization of each substrate is governed by a unique mechanism of control with β-catenin-dependent signaling regulating fatty acid β-oxidation, while glucose and glutamine utilization are β-catenin-independent and downstream of mammalian target of rapamycin complex 2 (mTORC2) and mammalian target of rapamycin complex 1 (mTORC1) activation, respectively. The emergence of these data has provided a new context for the mechanisms by which Wnt signaling influences bone development.
INTRODUCTION
The dynamic nature of bone tissue requires a homeostatic balance between new bone formation and the resorption of old or damaged matrix to maintain skeletal architecture and strength. The skeleton must balance the need to provide a rigid structure that can protect vital organs and facilitate locomotion against its function as a mineral reserve for the entire body. Osteoclastic cells, which degrade bone matrix and liberate the calcium and phosphate stored as hydroxyapatite, are derived from the hematopoietic lineage, while bone-forming osteoblasts responsible for the deposition and mineralization of new bone matrix are of mesenchymal origin. Understanding the local, hormonal, and genetic effectors that influence the activity of these two cell types is critical to our understanding of human disease and the development of new therapeutics that increase bone mass and strength [12].
In our aging population, the close association and often coexisting conditions of osteopenia, obesity, diabetes, and cancer have peaked an interest in the effects of intermediary metabolism on bone cell function. The initial analyses of fuel selection by bone cells were performed more than 50 years ago, were focused on the osteoblast and the role that metabolites might play in the liberation of mineral ions, but were forgotten by the field. Glucose was proposed as the primary energy source for the osteoblast and a carbon source for amino acid and collagen synthesis, while the metabolites citrate and lactate were expected to provide an acidic environment sufficient for the release of calcium from the bone matrix [34567]. The oxidation of palmitate, the most abundant fatty acid in animals, by osteoblasts was suggested to contribute between 40% and 80% of the energy produced by glucose oxidation [8]. More recently, observations made in genetic mouse models or in cell culture using more sophisticated bioenergetic analyses have provided confirmation of these classic studies and elaborated on the changes in metabolic flux that accompany each stage of osteoblast differentiation [910]. Glucose uptake via glucose transporter 1 (Glut1) is now recognized as a key regulator of the molecular events that initiate early osteoblast commitment by regulating the stability of Runt-related transcription factor 2 (Runx2) [11]. Likewise, studies using radiolabeled lipoproteins and fatty acids indicate that the skeleton plays a role in lipid homeostasis [1213].
Emerging evidence suggests that the utilization of specific fuel substrates by the osteoblast is governed by key developmental and hormonal signals [1415161718]. Key among these is the Wnt signaling pathway that is critical for normal bone mass accrual and exerts control of over nearly all facets of osteoblast maturation and function. In the sections below, we provide overviews of the Wnt signaling pathway and its role in whole body metabolism before describing the effect of Wnt signaling on fatty acid, glucose and glutamine catabolism by the osteoblast.
Wnt SIGNALING
Wnt signaling plays a central role in the coordination of a number of cellular and organismal processes including proliferation, tissue development and repair, and metabolism [192021]. The most thoroughly studied pathway, referred to as Wnt/β-catenin signaling or the “canonical” pathway, regulates the proteasomal degradation of the transcription factor, β-catenin [20]. In the absence of Wnt ligands, glycogen synthase kinase-3β (Gsk3β) and casein kinase-1 (Ck-1), in collaboration with a destruction complex that contains the adenomatous polyposis coli (Apc) protein [22], the Wilms tumor suppressor protein (WTX) [23], and Axin [2425], sequentially phosphorylate cytosolic β-catenin [26]. These modifications facilitate the recognition of β-catenin by β-transducing repeat-containing protein (β-TrCP), a component of an E3 ubiquitin ligase complex, and its targeting for degradation [2728]. Wnt ligands inhibit the proteolysis of newly synthesized β-catenin [29] by stimulating the formation of a multiprotein receptor complex composed of a seven transmembrane Frizzled receptor [3031] and a low-density lipoprotein related receptor-5 (Lrp5) or Lrp6 co-receptor [3233]. Ligand engagement leads to the phosphorylation of the intracellular domain of Lrp5 and Lrp6 [34], the recruitment of disheveled (Dvl) [353637] and Axin [38], and ultimately the cytoplasmic accumulation and then nuclear translocation of β-catenin. Within the nucleus, β-catenin regulates target gene expression by interacting with DNA-bound T-cell factor (TCF) [3940] and disrupting its association with the transcriptional repressor, Groucho [4142]. Transcriptional activity is further enhanced by the phosphorylation of β-catenin and TCF [4344] and the recruitment of co-activators and histone modifying enzymes that interact with the C- and N-terminal tails of β-catenin [454647].
The binding of Wnt ligands to Frizzled and Lrp5/6 can also initiate signaling downstream of the mammalian target of rapamycin complex 1 (mTORC1) and mammalian target of rapamycin complex 2 (mTORC2) complexes. Gsk3β, the kinase that phosphorylates β-catenin and targets it for degradation, phosphorylates the tuberous sclerosis 2 (Tsc2) protein at two serine residues to enhance its inhibition of mTORC1. Therefore, by inhibiting Gsk3β activation, Wnt ligands increase protein synthesis and cell growth [4849]. Activation of the mTORC2 complex by Wnts has not been as well studied, though Wnt3a, Wnt7b, and Wnt10b are able to stimulate its activity in osteoblastic cells [50] and the complex is required for the osteoanabolic effect of sclerostin neutralization [51]. In vitro gene knockdown studies indicated that the signaling mechanism involves the small GTPase, Rac family small GTPase 1 (Rac1) [50].
Other “non-canonical” pathways that do not activate β-catenin or require a Lrp5/Lrp6 co-receptor are also activated by the interaction of Wnt ligands with Frizzled receptors. These pathways predominately affect processes like cellular migration and polarity [525354] and their activation may antagonize the activation of Wnt/β-catenin signaling [55565758]. In the Wnt-Ca2+ pathway, Wnt stimulation induces calcium transients [585960] that activate calcium/calmodulin-dependent kinase II, calcineurin, and protein kinase C [6162]. In another pathway, known as Wnt-Frizzled planar cell polarity, Frizzled and the four transmembrane protein, Vangl, together with four other core proteins interact across cell membranes to regulate cellular directionality [5463]. The role of Wnt ligands in this pathway is less clear, but both Wnt-5a and Wnt-11 [646566] have been implicated in the process.
CONTRIBUTIONS OF Wnt SIGNALING TO SKELETAL HOMEOSTASIS
The concept that Wnt signaling regulates skeletal development and homeostasis was first evident in mouse mutants deficient or hypomorphic for Wnt3a [6768]. In these models, global disruption results in an axial truncation caudal to the forelimbs with a lack of somites and extensive death of mesodermal cells, while hypomorphic alleles lead to deficiencies in ossification with malformations and fusions of caudal vertebrae. However, the notion that Wnts are required for normal bone acquisition gained significant momentum when three publications linked mutations in the human LRP5 gene that encodes the Wnt co-receptor to conditions with high and low bone mass in humans. In 2001, Gong and colleagues [69] from the Osteoporosis Pseudoglioma (OPPG) Syndrome Collaborative Group reported that loss-of-function mutations in LRP5 were causal for the development of OPPG, a condition characterized by severe, early-onset osteoporosis as well as disruptions in ocular structure or the persistence of vitreal vascularization. Less than a year later, Little et al. [70] and Boyden et al. [71] independently identified mutations leading to a glycine-to-valine amino acid change (G171V) in LRP5 in kindreds with a high bone mass (HBM) phenotype. This missense mutation was revealed to inhibit the binding of dickkopf and sclerostin, two secreted Wnt signaling antagonists, to LRP5 thereby enhancing signaling capacity [7172737475]. Subsequent studies have identified additional mutations in LRP5 as well as LRP6 and other Wnt signaling components that influence bone mass and strength [7677787980].
Numerous transgenic mouse models have also now been created to examine the cellular and molecular basis by which Wnt signaling governs skeletal modeling/remodeling. Most of these models and especially mice globally deficient for Lrp5 and those expressing HBM alleles recapitulate the OPPG and HBM phenotypes, respectively [818283]. Wnt/β-catenin signaling is required for the initial fate specification of cells committing to the osteoblast lineage [8485], regulates the performance of maturing osteoblasts [828687], controls osteoclastogenesis [8889], and also influences responsivity of osteoblasts to anabolic hormones [90919293]. Dramatic examples of the central role of Wnt/β-catenin signaling in skeletal homeostasis are evident in the work of Holmen et al. [89] who generated mice in which the gene encoding β-catenin or the Apc protein were ablated specifically in the osteoblast. The β-catenin deficient mice developed severe osteopenia due to a reduction in osteoblast numbers and a dramatic increase in the prevalence of osteoclasts, while Apc mutants exhibited increased β-catenin activation and bone overgrowth. Strikingly, neither model was compatible with prolonged postnatal life. Osteocyte-specific β-catenin knockout mice (via expression of the dentin matrix protein 1 [DMP1]-Cre transgene) also have a severe skeletal phenotype that resembles the osteoblast-specific mutant models, with an expanded marrow cavity and thin cortical bone due to increased resorption by osteoclasts and changes in the osteoprotegerin (OPG)/receptor activator of nuclear factor kappa-B ligand (RANKL) ratio, but without a change in numbers or activity of osteoblasts [94]. Thus, it is possible that β-catenin actions in the osteocyte regulate the activity of osteoclasts, while osteoblastic β-catenin regulates cell maturation.
IMPACT OF Wnt SIGNALING ON WHOLE BODY METABOLISM
The notion that Wnt signaling contributes to the regulation of whole body metabolism was evident from the initial cloning of the gene encoding the LRP5 co-receptor in humans. Located on the q-arm of chromosome 11, LRP5 was identified as one of four genes in a 400 kb region surrounding the insulin dependent diabetes mellitus 4 (IDDM4) locus that exhibited strong genetic linkage with the development of type 1 diabetes [9596]. Subsequent studies would reveal that LRP5 was not the causative gene at this locus [97], but polymorphisms in LRP5 (A1330V, N740N, Q89R) have been linked to increased total and low-density lipoprotein cholesterol levels, hypertension, increased body mass index, and obesity [9899100101102]. Indeed, recent work from Loh et al. [103] reported that HBM mutations in LRP5 in humans are associated with a metabolically favorable body fat distribution and increased insulin sensitivity, while low bone mass, loss of function mutations are associated with increased abdominal fat accumulation.
The extracellular domain of LRP5 contains 3 low-density lipoprotein receptor (Ldlr) domains that retain the capacity to bind apolipoprotein E (ApoE) [104] and mouse studies performed by Fujino and colleagues [105] suggest a direct role for the protein in glucose and lipoprotein metabolism. On a standard chow diet, Lrp5−/− mice exhibit age-related impairments in glucose-stimulated insulin secretion which is likely due to alterations in glucose-stimulated ATP production and Ca2-transients. With high fat diet feeding, the mutants develop hypercholesterolemia secondary to a reduction in hepatic chylomicron clearance [105]. Crossing the Lrp5−/− mice onto an ApoE−/− background, to generate double mutants, results in hypercholesterolemia even on a chow diet and to a greater extent than ApoE-deficiency alone, as well as impaired fat tolerance, and advanced atherosclerosis [106]. Lrp5 appears, therefore, to contribute to lipoprotein metabolism in a pathway that works in parallel with the Ldlr.
Genome wide association studies have implicated other Wnt signaling components in the development of metabolic disease, with polymorphisms in WNT5B [107] and WNT10B [108] linked to the development of type 2 diabetes and obesity, respectively. More strikingly, Grant and colleagues [109] identified a strong association between variants in the gene encoding the Wnt effector transcription factor 7-like 2 (TCF7L2, also referred to as TCF4) with susceptibility for type 2 diabetes in an Icelandic cohort and then replicated the finding in Danish and American cohorts. Additional studies have now replicated this association in a number of other ethnic populations [110111112]. The expression of Tcf7l2 appears to be regulated by alterations in the metabolism of a number of tissues, including the pancreas, adipose tissue, and the liver [113114115]. Direct examination of the transcriptional regulator's mechanism of action were initially hampered by the perinatal death of Tcf7l2−/− mice [116], but heterozygous mice were shown to be protected from the development of a diabetic phenotype, while those that overexpressed the human gene exhibited an increased susceptibility when fed a high fat diet [117]. TCF7L2 was expected to exert its effect on metabolism via the β-cell and two groups demonstrated that loss of Tcf7l2 function in the pancreas of transgenic mouse models resulted in impaired glucose tolerance [118119]. However, tissue-specific knockouts generated by Boj and colleagues [120], in which Tcf7l2 expression was ablated in either the pancreatic β-cell or the hepatocyte, suggested a different mechanism of action. Their β-cell-specific Tcf7L2 knockouts exhibited normal islet development and function, but hepatocyte-specific ablation reduced glucose production and improved glucose homeostasis. These results remain controversial and the mechanisms of TCF7L2 actions in metabolism continue to be an area of intense interest [121]. It is likely that the actions of TCF7L2 in metabolic control represent combinatorial effects across a number of tissues.
Wnt-STIMULATED β-OXIDATION IN THE OSTEOBLAST
Our group's interest in the metabolic actions of Wnt signaling in the skeleton stems from a serendipitous observation made during an evaluation of the unique and overlapping functions of the Wnt co-receptors, Lrp5 and Lrp6, in the osteoblast. Consistent with the contributions of Lrp5 and Lrp6 to the activation of β-catenin-dependent signaling and the roles of Wnt signaling in regulating osteoblast function noted above, genetic ablation of either Wnt co-receptor in the mature osteoblast (Lrp5flox; Oc-Cre or Lrp6flox; Oc-Cre) impaired skeletal homeostasis and resulted in the development of an osteopenic phenotype [82]. Surprisingly, Frey et al. [122] demonstrated that the Lrp5 mutants also developed alterations in body composition that were not evident in the Lrp6 mutants. Loss of Lrp5 function increased the size of white adipose tissue depots, reduced whole body energy expenditure indexed by indirect calorimetry, and resulted in the development of dyslipidemia marked by increased levels of serum triglycerides and free fatty acids. Subsequent gene expression profiling of Lrp5-deficient osteoblasts cultured in vitro suggested that the phenotype was a result of altered fatty acid catabolism, as the expression of a number of genes involved in mitochondrial long-chain fatty acid oxidation were down-regulated in the mutant osteoblasts (Fig. 1). Indeed, Lrp5-deficient osteoblasts exhibited an impaired ability to fully oxidize oleate to CO2.
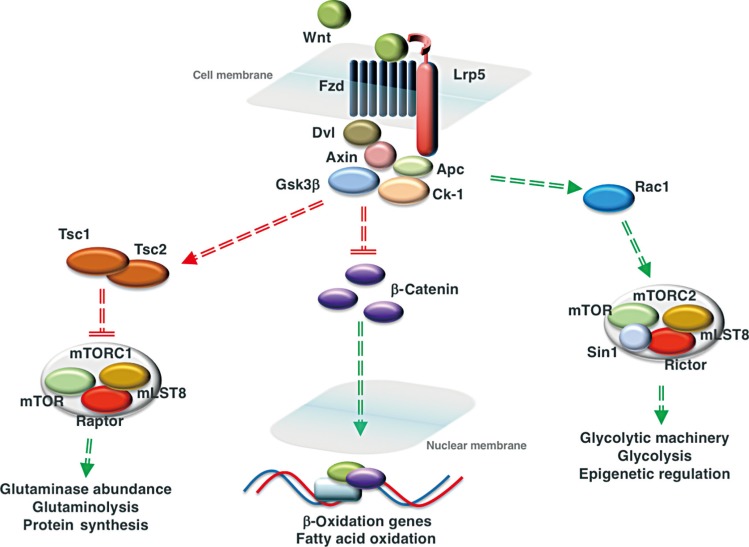
Wnt signaling regulates the utilization of three fuel substrates by cells of the osteoblast lineage. Activation of Wnt signaling via the interaction of a Wnt ligand with a frizzled receptor (Fzd) and the low-density lipoprotein related receptor-5 (Lrp5) co-receptor inactivates the destruction complex consisting of disheveled (Dvl), Axin, glycogen synthase kinase-3β (Gsk3β), adenomatous polyposis coli (Apc), and casein kinase-1 (Ck-1). In mature osteoblasts, this allows the accumulation of β-catenin and its translocation to the nucleus, where the transcription factor activates the expression of genes involved in mitochondrial long-chain fatty acid oxidation. In osteoprogenitors, Wnt signaling activates mammalian target of rapamycin complex 1 (mTORC1) and mammalian target of rapamycin complex 2 (mTORC2) signaling to increase glutaminolysis and glycolysis, respectively. Wnt ligand binding inhibits Gsk3β activity and its ability to activate the tuberous sclerosis 1/2 (Tsc1/Tsc2) complex that inhibits mTORC1 activity. Activation of mTORC1 increases the abundance of glutaminase, the first enzyme in glutaminolysis. Activation of the mTORC2 complex, which regulates the abundance of proteins involved in glycolysis, is downstream of Rac family small GTPase 1 (Rac1). By inhibiting the entry of glucose into the tricarboxylic acid (TCA) cycle, Wnt regulates the availability of substrates for histone acetyltransferases. Red lines represent interactions that are suppressed by the activation of Wnt signaling, while green lines indicate interactions that are enhanced. mLST8, mammalian lethal with SEC13 protein 8; Sin1, stress activated protein kinase interacting protein 1.
In a follow-up study, Frey et al. [123] investigated whether the regulation of long-chain fatty acid oxidation by Lrp5 required the activation of β-catenin. Initially, cultures of primary osteoblasts were treated with Wnt ligands expressed in the bone microenvironment [124125126] and their ability to influence β-oxidation was assessed. Only ligands like Wnt3a, Wnt10b and Wnt16 that are able to induce β-catenin activation enhanced oleate oxidation, which suggested that Wnt-induced alterations in metabolism proceeded via the canonical mechanism. As a more direct test of this hypothesis, Frey et al. [123] generated mice in which the genetic ablation of the catenin beta 1 (Ctnnb1) gene in the osteoblast could be controlled by the administration of tamoxifen (Ctnnb1flox; Oc-CreERT2) to avoid the early lethality associated with constitutive disruption of β-catenin expression in this cell population [89]. In vitro, the loss of β-catenin function in cultures of maturing osteoblasts resulted in the expected inhibition of osteoblast maturation as well as a nearly 50% reduction in the capacity for oleate oxidation and a significant reduction in cellular ATP content, despite an increase in glucose uptake and glycolytic metabolism. In vivo, manipulation of β-catenin expression mirrored the effect of Lrp5 loss of function as the mutants developed an increase in adipose tissue mass and an increase in serum fatty acids. Surprisingly, the β-catenin mutants also developed impairments in glucose tolerance and insulin sensitivity that were not evident in Lrp5 mutants and are likely secondary to ectopic lipid accumulation. The more severe metabolic phenotype that develops in the β-catenin mutants is likely a result of an impairment in all effects of canonical Wnt signaling with the deletion of the pathway's target transcription factor, while the presence of Lrp6 may be able to partially compensate and maintain a level of Wnt signaling in Lrp5 mutants.
To explore the absolute requirement for fatty acid catabolism during bone development and to determine if bone contributes to whole body lipid homeostasis, Kim et al. [13] examined the skeletal and metabolic phenotypes of a mouse model in which the expression of carnitine palmitoyltransferase 2 (Cpt2), an obligate enzyme for mitochondrial fatty-acid β-oxidation, was disrupted in osteoblasts and osteocytes. Consistent with the observation that bone takes up a significant quantity of circulating fatty acids, skeletal homeostasis was impaired by Cpt2 deficiency, but the severity was dependent on the sex of the mutant. Male Cpt2 mutants exhibited only a transient decrease in trabecular bone volume that was most evident at 6 weeks of age, but female mutants failed to reach peak trabecular bone volume and exhibited a decrease in trabecular bone volume in both the distal femur and L5 vertebrae at all timepoints examined after 1 month of age. Cortical tissue area in the femur of female mice was increased although cortical thickness did not change, implying that the skeleton adapted to reduced bone quality by changing geometrical properties of the femur to maintain bone strength. Histomorphometric analyses revealed that the skeletal phenotype in the mutant mice was secondary to a mineralization defect, as the female mutants accumulated unmineralized matrix and exhibited a reduction in the mineral apposition rate and increase in the mineralization lag time. A combination of in vivo and in vitro studies suggested that the sexually dimorphic phenotype is related to the ability of estrogen to influence adjustments in fuel selection. Glucose uptake was increased in Cpt2 deficient osteoblasts cultured in vitro and the skeletal tissue of male Cpt2 deficient mice, but not in the bone of female mutants or in mutant osteoblast cultures treated with exogenous estrogen. Estrogen treatment of primary cultures of mutant osteoblasts also exacerbated the downregulation of genes associated with osteoblast differentiation and resulted in a more severe impairment in matrix mineralization. A similar influence of estrogen on cellular metabolism has been noted in a number of other tissues [127128129].
Like genetic ablation of Lrp5 or β-catenin, inhibition of long-chain fatty acid metabolism in the osteoblast resulted in an increase in serum fatty acids, but on a normal chow diet the male Cpt2 mutants exhibited a reduction in body fat fraction and in the weight of the gonadal fat pad. This body composition phenotype is likely due to a shift in glucose utilization and storage because glucose uptake by adipose was repressed while skeletal glucose uptake was increased. Intriguingly, when the male Cpt2 mutants were fed a high fat diet that increased the levels of estrogen, bone loss ensued, the weights of all major fat pads increased, and the mutant mice performed poorly in glucose tolerance and insulin tolerance tests. Overall, these data demonstrated that fatty acid catabolism is required for normal osteoblast function and bone mass acquisition and is strongly influenced by both sex and diet.
Wnt-STIMULATED GLYCOLYSIS IN THE OSTEOBLAST
As indicated above, glucose is required for normal osteoblast function and likely represents an important energy source. Since components of the Wnt signaling cascade have been linked to the regulation of whole-body glucose metabolism, Esen and colleagues [50] explored the effect of Wnt signaling on glucose utilization by osteoblasts. Relying primarily on the ST2 cell line, which models mouse bone marrow stromal cells, Wnt3a and Wnt10b were demonstrated to increase glucose acquisition by stimulating the expression of Glut1, hexokinase-2, lactate dehydrogenase, and pyruvate dehydrogenase kinase 1 and were more effective than high dose insulin stimulation. Intriguingly, the increase in glucose uptake was not accompanied by an increase in the oxygen consumption rate, which suggests that it was not processed via oxidative phosphorylation. Rather, Wnt3a stimulated lactate production and increased the extracellular acidification rate, suggesting that Wnt signaling activated aerobic glycolysis, a metabolic process most closely associate with cancer cell metabolism [130].
Using pharmacological antagonists and gene knockdown studies to elaborate on the mechanism by which Wnts stimulate glycolysis, Esen et al. [50] demonstrated that the response required the Wnt co-receptor Lrp5. Indeed, Lrp5−/− mice as well as those in which Lrp5 expression was abolished via osterix-Cre expression exhibited a reduction in glycolytic enzyme expression in bone. However, it did not proceed via alterations in the activity of either Gsk3β or β-catenin activity, as inhibiting the activity of these effectors did not impact glucose consumption. Instead, Wnt3a activated mTORC2 via Rac1, which in turn coordinated the increase in glucose consumption and the increase in glycolytic gene expression.
Why Wnt signaling, which has profound anabolic effects on osteoblast differentiation and function, should lead early osteoblasts to rely on a less efficient mode of ATP generation from glucose remains an open question. One possibility is that like cancer cells, the products of aerobic glycolysis are used by immature cells as the starting material for biosynthetic pathways that produce amino acids and nucleotides [130]. A second possibility suggested by the Long Laboratory is that this metabolic paradigm contributes to the epigenetic regulation of osteoblast differentiation [131]. Using RNA sequencing, Karner et al. [131] demonstrated that the number of genes exhibiting an increase in expression after Wnt3a stimulation of ST2 cells was surpassed by the number of genes exhibiting a decrease in expression and that many of the downregulated genes were associated with differentiation toward the chondrocyte or adipocyte lineages (including peroxisome proliferator-activated receptor gamma [Pparg] and CCAAT enhancer binding protein alpha [Cebpa]). Since gene activation is expected to be β-catenin's major mode of action, the group postulated that Wnts contributed to gene suppression via the regulation of histone modification. Consistent with this idea, bulk histone acetylation was reduced in ST2 cells after Wnt3a treatment, but the activity levels of histone deacetylases and histone acetyltransferases were not affected. Instead, Wnt signaling reduced the availability of the histone acetyltransferase substrate, acetyl coenzyme A (acetyl-CoA), as well as its precursor, citrate. Therefore, by inhibiting the entry of glucose into the tricarboxylic acid (TCA) cycle and thereby reducing citrate and acetyl-CoA production, Wnt signaling regulates the genomic landscape and fate-specification of osteoprogenitors.
Wnt-STIMULATED GLUTAMINOLYSIS IN THE OSTEOBLAST
The amino acid, glutamine, represents a third energy source whose catabolism is under the control of Wnt signaling in the osteoblast. Glutamine is abundant in the circulation (approximately 20% of the free amino acid pool) and in the neoplastic cells and normal cells (i.e., lymphocytes, fibroblasts, etc.) that utilize Warburg-type metabolism it can be utilized in a process referred to as anaplerosis. This process maintains TCA function when intermediates like citrate are removed from the cycle for anabolic reactions. In this reaction, glutaminase, a mitochondrial enzyme, deaminates glutamine to form glutamate which is then converted to α-ketoglutarate, a TCA intermediate, by glutamate dehydrogenase or via transamination with pyruvate by alanine aminotransferase. Biltz and colleagues [132] demonstrated more than 30 years ago that rat calvarial cells oxidize glutamine while more recent in vitro studies have suggested a requirement for glutamine supplementation for mineralization of bone matrix by osteoblasts [133].
Karner and colleagues [134] examined glutamine metabolism in the context of Wnt-stimulated osteoblast differentiation and the coordinate increases in protein synthesis necessary to prepare bone matrix. Wnt stimulation of the ST2 cell line increased glutamine uptake, but it was quickly metabolized, as the cells exhibited a nearly 40% decrease in cellular glutamine levels 24 hours after treatment due to an increase in glutaminase activity and the entry of glutamine-derived carbons into the TCA cycle via anaplerosis. Using pharmacological inhibition of glutaminase activity, the group demonstrated that glutamine catabolism was required for Wnt-stimulated osteoblast differentiation in vitro as well as the increase in bone mass resulting from the expression of a HBM variant of the Lrp5 co-receptor in vivo. In parallel, glutaminolysis initiated the activation of the integrated stress response and the associated increase in the expression of genes necessary for amino acid uptake and protein folding, which were also required for Wnt-stimulated bone acquisition. Consistent with this close association with protein synthesis, mechanistic studies indicated that the regulation of glutamine utilization is dependent on the activation of mTORC1. Thus, glutamine acquisition and utilization in response to Wnt signaling appear to represent a molecular rheostat that acts to maintain cellular energetics, endoplasmic reticulum status [134] and redox balance [17].
CONCLUSIONS
Taken together the studies reviewed above highlight an exciting and newly appreciated effect of Wnt signaling on the biology of the osteoblast. This evolutionarily-conserved pathway that is critical for the attainment of normal bone mass and structure coordinates one of the most essential cellular functions: the generation of energy necessary to fuel other cell processes. The existing data suggest that the governance of osteoblast fuel selection by Wnt is highly dependent on the state of osteoblast differentiation, with immature osteoblasts exhibiting an increase in glucose and glutamine utilization and mature cells switching to and increasing the utilization of fatty acids. These findings accord with well-established concepts in developmental biology and the changing metabolic demands of osteoblast differentiation [9]. Differentiated osteoblasts that prepare and mineralize the bone matrix maintain abundant mitochondria likely as a result of the tremendous energetic demands of protein synthesis [135136]. It follows that this stage of cellular differentiation is associated with fatty acid β-oxidation, which has the capacity to produce approximately 131 ATP per molecule of palmitate, in response to anabolic Wnt stimulation. Immature bone cells instead seek to maintain the redox balance, checks on endoplasmic reticulum status, and substrates for epigenetic regulation and increasing cellular biomass offered by glycolysis and glutaminolysis as Wnt signaling stimulates their commitment to the osteoblast lineage.
Additional basic and translational studies are necessary to further probe the contributions of Wnt signaling to osteoblast metabolism and how this contributes to global energy balance. The sheer size of the skeleton and its cellular biomass suggests that anabolic signals like Wnt should lead to the reallocation of energy sources. Indeed, models of Lrp5 deficiency in bone exhibited changes in serum lactate and lipids. It is also likely that Wnt contributes to the regulation of mitochondrial biogenesis. This interaction has been examined in other tissues [137138], but not yet in the osteoblast. Finally, while compelling evidence for the role of Wnt signaling in whole body metabolism and the regulation of skeletal dynamics exists in humans, data on the effects of Wnt on the intermediary metabolism of human osteoblasts is still lacking. Such data should be an essential part of our understanding of Wnts actions in the skeleton as therapeutics that target the Wnt pathway enter the clinic.
ACKNOWLEDGMENTS
The authors are grateful to the many other investigators whose work has not been cited here due to space limitations. This work was supported by National Institutes of Health grant DK099134 and Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development grant BX003724.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.