Mitochondrial Complexes I and II Are More Susceptible to Autophagy Deficiency in Mouse β-Cells
Article information

Abstract
Background
Damaged mitochondria are removed by autophagy. Therefore, impairment of autophagy induces the accumulation of damaged mitochondria and mitochondrial dysfunction in most mammalian cells. Here, we investigated mitochondrial function and the expression of mitochondrial complexes in autophagy-related 7 (Atg7)-deficient β-cells.
Methods
To evaluate the effect of autophagy deficiency on mitochondrial function in pancreatic β-cells, we isolated islets from Atg7F/F:RIP-Cre+ mice and wild-type littermates. Oxygen consumption rate and intracellular adenosine 5'-triphosphate (ATP) content were measured. The expression of mitochondrial complex genes in Atg7-deficient islets and in β-TC6 cells transfected with siAtg7 was measured by quantitative real-time polymerase chain reaction.
Results
Baseline oxygen consumption rate of Atg7-deficient islets was significantly lower than that of control islets (P<0.05). Intracellular ATP content of Atg7-deficient islets during glucose stimulation was also significantly lower than that of control islets (P<0.05). By Oxygraph-2k analysis, mitochondrial respiration in Atg7-deficient islets was significantly decreased overall, although state 3 respiration and responses to antimycin A were unaffected. The mRNA levels of mitochondrial complexes I, II, III, and V in Atg7-deficient islets were significantly lower than in control islets (P<0.05). Down-regulation of Atg7 in β-TC6 cells also reduced the expression of complexes I and II, with marginal significance (P<0.1).
Conclusion
Impairment of autophagy in pancreatic β-cells suppressed the expression of some mitochondrial respiratory complexes, and may contribute to mitochondrial dysfunction. Among the complexes, I and II seem to be most vulnerable to autophagy deficiency.
INTRODUCTION
Autophagy is a cellular degradation-recycling system for aggregated proteins and damaged organelles. Autophagy to remove damaged mitochondria is termed mitophagy. Impairment of autophagy is known to induce the accumulation of damaged mitochondria and to cause mitochondrial dysfunction in most mammalian cells. The accumulation of deformed mitochondria has been noted in hepatocytes and cardiomyocytes of autophagy- deficient mice [1,2]. Autophagy-related 7 (Atg7), an E1-like gene, is essential for the formation of autophagosomes [3], and Atg7-deficient erythrocytes show altered mitochondrial membrane potential [4]. This suggests that mitophagy plays a pivotal role in the control of mitochondrial quality and quantity [5].
Recently, autophagy has also been implicated in the regulation of pancreatic β-cell function and mass [6]. Mice with β-cell-specific deletion of Atg7 showed impaired glucose tolerance and decreased serum insulin levels [7,8]. In those studies, electron microscopy of the autophagy-deficient β-cells revealed mitochondrial swelling. Other groups have reported decreased oxygen consumption rate and adenosine 5'-triphosphate (ATP) production in Atg7-deficient islets ex vivo, suggesting mitochondrial dysfunction [8,9]. In addition, reduced Ca2+ transients in response to glucose in autophagy-deficient β-cells have been noted [7], which may indicate abnormal mitochondrial function. Mitochondria are crucial in the generation of ATP, affecting both the closure of ATP-sensitive K+ channels and Ca2+ influx. Here, we further investigated changes in Atg7-deficient β-cells, focusing on the level of the mitochondrial respiratory chain complexes.
METHODS
Mice and islet isolation
To generate mice with β-cell-specific deletion of Atg7 (Atg7Δβ-cell), Atg7-floxed (Atg7F/F) mice were crossed with RIP-Cre mice. RIP-Cre mice or Atg7F/F mice were used as controls. After overnight fasting, pancreatic islets were isolated from 20-week-old mice using a collagenase digestion technique [7]. All animal experiments were conducted in accordance with the Institutional Guide for the Care and Use of Laboratory Animals of Seoul National University Hospital.
Cells and Atg7 RNA interference
β-TC6 cells were obtained from ATCC (Manassas, VA, USA). The cells were cultured in Dulbecco's Modified Eagle's Medium (GIBCO, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin (GIBCO), in a humidified atmosphere of 95% air and 5% CO2. To generate Atg7-deficient cells, β-TC6 cells were transfected with siAtg7 by electroporation using an Amaxa Cell Line Nucleofector Kit V system (Lonza, Basel, Switzerland). The glucose concentration of the culture medium was changed to 15 mM during the 48 hours of transfection. Down-regulation of Atg7 was verified by quantitative real-time polymerase chain reaction (RT-PCR).
Oxygen consumption rate
Baseline oxygen consumption rate was measured from 200 islets over 2 hours, using a BD Oxygen Biosensor System, according to the manufacturer's protocol (Becton Dickinson, San Jose, CA, USA). An Oxygraph-2k (OROBOROS Instruments, Innsbruck, Austria) was used to examine the responses to mitochondrial effectors of 1,000 islets suspended in Krebs-Ringer bicarbonate HEPES (KRBH) buffer with 10 mM glucose, at 37℃. During the experiments, substances were added serially as described [10,11]: glutamate (10 mM) and malate (2 mM) (G+M) were first added as substrates for mitochondrial respiration; adenosine diphosphate (ADP, 2.5 mM) was then added to induce state 3 respiration; cytochrome c (10 µM) was added to test the integrity of the outer mitochondrial membrane; succinate (10 mM) was added for electron transfer to complex II; next, 0.5 to 1.5 µM carbonyl-cyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP), a mitochondrial respiration uncoupler, was added to obtain maximal oxygen consumption rate; rotenone (0.5 µM) was added to inhibit complex I, and then antimycin A (1.5 µM) to inhibit complex III.
Intracellular ATP content
Ten isolated islets in each of five culture wells were treated with low glucose-KRBH buffer (1.6 mM) or high glucose-KRBH buffer (16 mM) for 1 hour, after 1 hour of fasting. ATP content of the islets was then measured, using a bioluminescence assay kit (Sigma-Aldrich, St. Louis, MO, USA), and the values were normalized to total cellular protein in each sample. Protein concentration was determined using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA).
Real-time quantitative reverse transcription polymerase chain reaction of mitochondrial complex mRNAs
We isolated total RNA from mouse islets or β-TC6 cells using TRizol reagent (Ambion, Foster City, CA, USA), synthesized cDNA using M-MLV reverse transcriptase (Promega, Madison, WI, USA), and amplified it using the SYBR Premix Ex Taq polymerase (TaKaRa, Otsu, Japan). Quantitative RT-PCR was performed and analyzed using a LightCycler 96 Real-Time PCR System (Roche, Mannheim, Germany). Primer sequences are presented in Table 1.
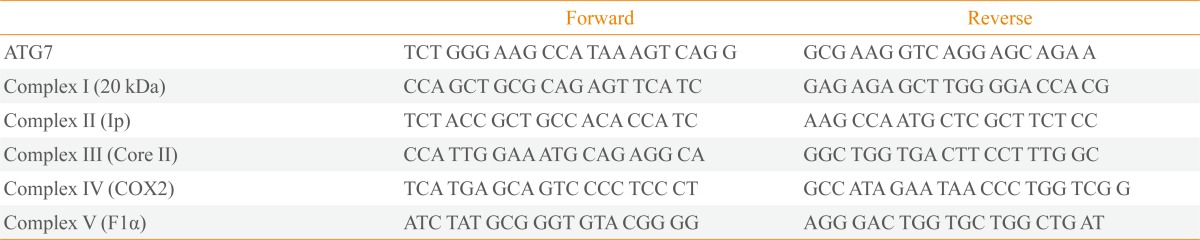
Sequences of Primers for Quantitative Real-Time Polymerase Chain Reaction
Statistical analysis
Results are presented as means±standard error. Statistical analyses of differences between groups were performed using Student t test or the Mann-Whitney U test, depending on sample variances. P values <0.05 were considered to indicate statistical significance.
RESULTS
Basal oxygen consumption rate
To investigate the effects of autophagy on mitochondrial function, islets were isolated from Atg7Δβ-cell and control mice, and oxygen consumption rate was measured ex vivo for 2 hours (Fig. 1). Oxygen consumption rate during that time increased 3-fold over initial values in control islets, while that of the Atg7Δβ-cell islets changed nonsignificantly. This difference between the groups in fold change after 2 hours was significant (P<0.05).
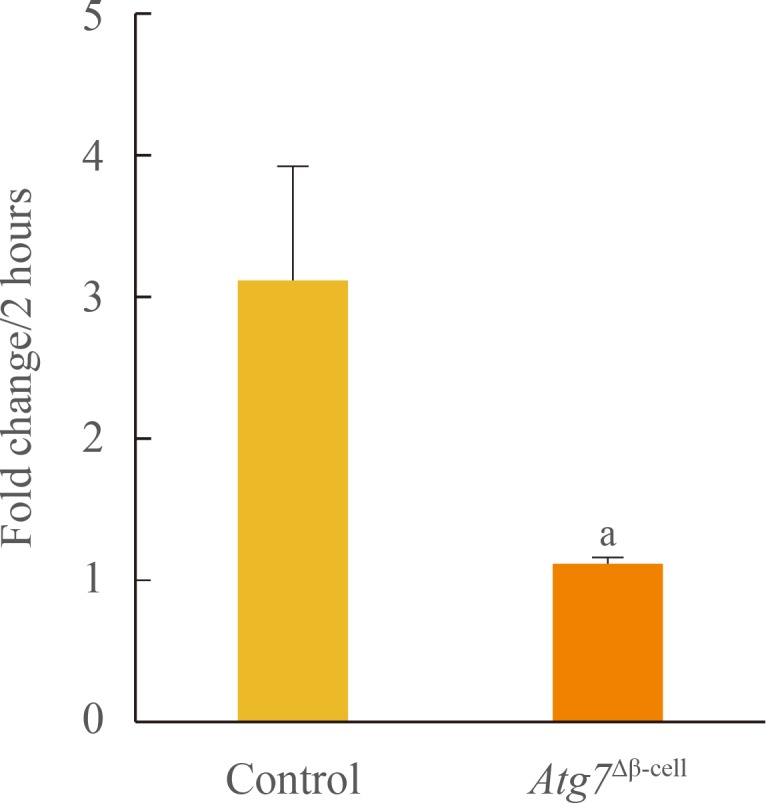
Basal oxygen consumption rates in islets isolated from Atg7Δβ-cell and control mice (n=5 to 8 per group). Data are presented as fold changes±SE, and comparisons were performed using a Mann-Whitney U test. Atg7, autophagy-related 7. aP<0.05 compared to the control.
Intracellular ATP content
We measured intracellular ATP content ex vivo in response to glucose stimulation. The intracellular ATP content of islets from Atg7Δβ-cell mice was significantly lower than that from control mice, under both low- and high-glucose conditions (P<0.05) (Fig. 2A). However, the fold change in ATP production with high glucose was comparable between the two groups (Fig. 2B). This may be due to suppressed ATP production under both low- and high-glucose concentrations.
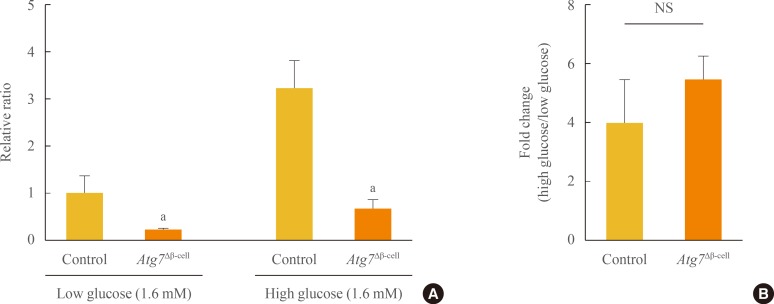
Intracellular adenosine 5'-triphosphate (ATP) content induced by glucose stimulation of islets isolated from Atg7Δβ-cell and control mice (n=3 to 10 per group). (A) ATP content under low-glucose (1.6 mM) and high-glucose (16 mM) conditions. All values were normalized relative to those of control islets under low glucose conditions. (B) Fold change in ATP content induced by high-glucose stimulation. Data are presented as fold changes±SE, and comparisons were performed using a Mann-Whitney U test. Atg7, autophagy-related 7; NS, not significant. aP<0.05 compared to control.
Mitochondrial respiration
It is unclear whether mitochondrial dysfunction reflects only the accumulation of damaged mitochondria or if there are other, direct effects on mitochondrial function. To determine whether specific respiratory states are influenced, we performed Oxygraph-2k analyses with isolated islets. Representative graphs are presented in Fig. 3A. Throughout the experiment, a marked reduction in oxygen consumption rate was observed in Atg7Δβ-cell islets compared to controls (P<0.01 by repeated measures analysis of variance) (Fig. 3B). Inhibitory effects of succinate (an inhibitor of complex II) and rotenone (an inhibitor of complex I) were relatively prominent, while there was no difference in responses to ADP (stage 3 respiration) or to antimycin A (an inhibitor of complex III). These findings suggest that complexes I and II are most vulnerable to autophagy deficiency.

Mitochondrial respiration in islets isolated from Atg7Δβ-cell and control mice (n=9 to 10 per group). (A) Representative graphs of mitochondrial respiration using Oxygraph-2k. Red lines indicate oxygen consumption rate in response to sequential loading of mitochondrial effectors (indicated by arrows below the graphs). (B) Quantitative comparisons of oxygen consumption rate in response to effectors. Data are presented as means±SE, and comparisons were performed using repeated measures analysis of variance and Mann-Whitney U tests. G+M, glutamate and malate; ADP, adenosine diphosphate; FCCP, carbonyl-cyanide-4-(trifluoromethoxy)-phenylhydrazone; AA, antimycin A; Atg7, autophagy-related 7. aP<0.05 compared to control islets.
Expression of mitochondrial complex genes
We examined the expression of mitochondrial respiratory complex genes in isolated islets and in β-TC6 cells. In Atg7Δβ-cell islets, expression of Atg7 mRNA was reduced by half (Fig. 4A). Among mitochondrial complexes, mitochondrial complex IV gene expression predominated. The expressions of mitochondrial complexes except complex IV were significantly lower in Atg7Δβ-cell islets than in controls (P<0.05) (Fig. 4A). In β-TC6 cells transfected with siAtg7 for 48 hours, Atg7 expression was also reduced by half (P<0.05); the expression of mitochondrial complexes I and II was reduced, but the statistical significance of this effect was marginal (Fig. 4B).
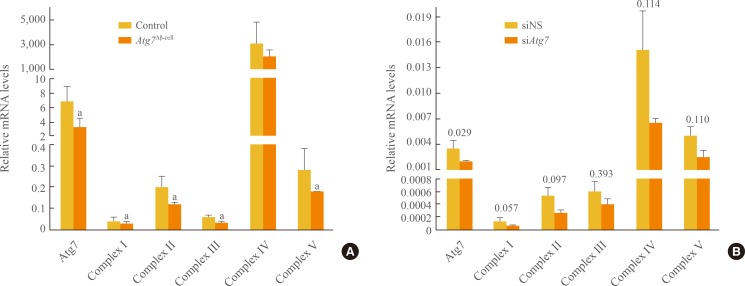
Expression of mitochondrial complex genes by quantitative real-time polymerase chain reaction (A) in Atg7Δβ-cell and control islets (n=7 to 8 per group), and (B) in β-TC6 cells transfected with siAtg7 or with siNS as a control (n=4). The expression levels were normalized to that of glyceraldehyde 3-phosphate dehydrogenase. Data are presented as means±SE, and comparisons were performed using Student t tests or Mann-Whitney U tests, depending on sample variances. P value is presented above the bar graph. Atg7, autophagy-related 7. aP<0.05 compared to control islets.
DISCUSSION
In pancreatic β-cells, as in other mammalian cells, damaged mitochondria are removed by autophagy [12]. Impairment of autophagy is thus expected to result in the accumulation of damaged mitochondria, and we have previously reported abnormal swollen mitochondria in Atg7Δβ-cell mice [7]. In this study, we observed the decreased oxygen consumption rate and ATP content, noted previously in autophagy-deficient pancreatic β-cells [8,9]. We also report that impaired autophagy affects specific steps in the mitochondrial electron transfer system. Although Atg7Δβ-cell islets showed a significant overall decrease in mitochondrial respiration compared to control islets (Fig. 3A), state 3 respiration estimated by treatment with ADP was unaffected by autophagy deficiency, and treatment with antimycin A, an inhibitor of complex III, abolished the difference in oxygen consumption rate between Atg7Δβ-cell islets and controls (Fig. 3B). Given the results of the significant differences by succinate and rotenone treatments, mitochondrial complexes I and II seemed to be most vulnerable to autophagy deficiency. Wu et al. [9] reported that Atg7-deficient mouse islets showed decreased mitochondrial respiration when treated with oligomycin, an inhibitor of complex V (ATP synthase), or with FCCP.
We evaluated mRNA expression of genes for components of each mitochondrial complex, both in mouse islets and in β-TC6 cells. Although Wu et al. [9] reported no difference in expression of these genes in Atg7-deficient skeletal muscle, we found the expression of all but one complex IV component gene to be decreased significantly in Atg7Δβ-cell islets (Fig. 4A). Inhibition of Atg7 in β-TC6 cells in vitro also reduced mitochondrial complex expression, although this effect was generally not statistically significant (Fig. 4B). The reduction in mitochondrial complexes I and II was marginally significant (P=0.057 and P=0.097, respectively). We presume that autophagy inhibition initially affects the expression of complexes I and II, and that prolonged suppression of autophagy influences expression of complexes III and V, but not IV. Such differences in gene expression would contribute to differences in the functions of the complexes.
Although we did not investigate reactive oxygen species (ROS), it may also contribute to mitochondrial dysfunction in autophagy-deficient β-cells. Mitochondria are the primary organelle producing ROS, and are also significantly influenced by ROS. As inhibition of Atg7 has been reported to increase the generation of ROS in pancreatic β-cells, and the attenuation of ROS recovered cellular function [9], mitochondrial dysfunction in autophagy-deficient β-cells may be closely related to ROS generation. In this study, we determined that autophagy impaired mitochondrial function. On the other hand, mitochondrial damage can generate ROS, leading to autophagy induction [13]. Rotenone, an inhibitor of complex I, and theonoyltrifluoroacetone (TTFA), an inhibitor of complex II, induced autophagic cell death, and inhibition of Atg5 ameliorated rotenone- or TTFA-induced cell death in transformed and cancer cell lines [14].
In conclusion, impairment of autophagy in β-cells in vivo induced mitochondrial dysfunction, particularly of mitochondrial respiratory complexes I and II. This suggests that not only the simple accumulation of damaged mitochondria but also direct effects on mitochondria, including altered expression of respiratory complex genes, may cause disturbed energy homeostasis by autophagy deficiency.
ACKNOWLEDGMENTS
This study was supported by a grant from the Innovative Research Institute for Cell Therapy (A062260) of the Ministry of Health and Welfare, Republic of Korea, and by a grant from Seoul National University Hospital (03-2010-0300).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.