Transcriptional Regulation of Fibroblast Growth Factor 21 Expression
Article information
Abstract
Fibroblast growth factor 21 (FGF21) is an attractive target for treating metabolic disease due to its wide-ranging beneficial effects on glucose and lipid metabolism. Circulating FGF21 levels are increased in insulin-resistant states; however, endogenous FGF21 fails to improve glucose and lipid metabolism in obesity, suggesting that metabolic syndrome is an FGF21-resistant state. Therefore, transcription factors for FGF21 are potential drug targets that could increase FGF21 expression in obesity and reduce FGF21 resistance. Despite many studies on the metabolic effects of FGF21, the transcriptional regulation of FGF21 gene expression remains controversial and is not fully understood. As the FGF21 transcription factor pathway is one of the most promising targets for the treatment of metabolic syndrome, further investigation of FGF21 transcriptional regulation is required.
INTRODUCTION
The fibroblast growth factor 21 (FGF21) family includes 22 members that are divided into seven subfamilies based on phylogeny and sequence [1,2]. Most FGF family members bind to FGF receptors on the cell surface and require heparin sulfate to stabilize the binding [3]. While classic FGFs act through an autocrine or paracrine mechanism to regulate cell growth and differentiation, FGF19 subfamily members including FGF15/19, FGF21, and FGF23 lack heparin-binding properties and can; therefore, be released into the circulation to act as endocrine factors [4]. In place of heparin, the transmembrane protein Klotho is required for FGF19 subfamily members to activate FGF receptors [5]; α-Klotho serves as a coreceptor for FGF23, while β-Klotho serves as a coreceptor for FGF15/19 and FGF21 [6,7,8,9].
FGF21 is a metabolic regulator that has favorable metabolic effects on glucose and lipid metabolism [10,11,12]. As mentioned above, interaction with β-Klotho is an essential step in FGF21-receptor complex activation. β-Klotho is almost exclusively expressed in the liver, adipose tissue, and pancreas [5], which may explain why these specific tissues are the predominant site of FGF21 action, although almost all tissues express FGF receptors [13]. FGF21 has attracted attention since Kharitonenkov et al. [10] discovered its potent insulin-sensitizing actions through increased glucose uptake in rodents. It is particularly important to distinguish between the systemic pharmacological effects of FGF21 during obesity and comorbid conditions and the tissue-specific effects of FGF21 that occur under more physiological conditions (Fig. 1). The physiological actions of FGF21 occur at lower concentrations and in more restricted organ systems and tissues than the pharmacological actions [14]. In fasting, FGF21 expression is induced by peroxisome proliferator-activated receptor α (PPARα) in the liver [15,16] and acts through endocrine mechanisms for adaptive starvation responses including gluconeogenesis, ketogenesis, torpor, and inhibition of somatic growth [12,14,17]. In the fed state, FGF21 expression is induced by PPARγ in white adipose tissue and acts in an autocrine or paracrine fashion to increase PPARγ activity [14,18,19,20]. As a consequence, during feeding, the induction of FGF21 in white adipose tissue fails to increase circulating levels of FGF21 [21]. Pharmacological administration of FGF21 affects multiple organs and tissues including the pancreas, adipose tissue, liver, and the central nervous system [14]. Systemic administration of FGF21 increases insulin sensitivity and energy expenditure, causing a loss of body weight and improvements in glucose and lipid metabolism in obese rodents and primates [10,11,22]. Furthermore, several reports have also shown that administration of FGF21 results in a significant decrease in lipid accumulation in the liver of diet-induced obese mice [22], suggesting that FGF21 could be a promising drug candidate for treatment of metabolic syndrome, with benefits for many of the symptoms of this disease.
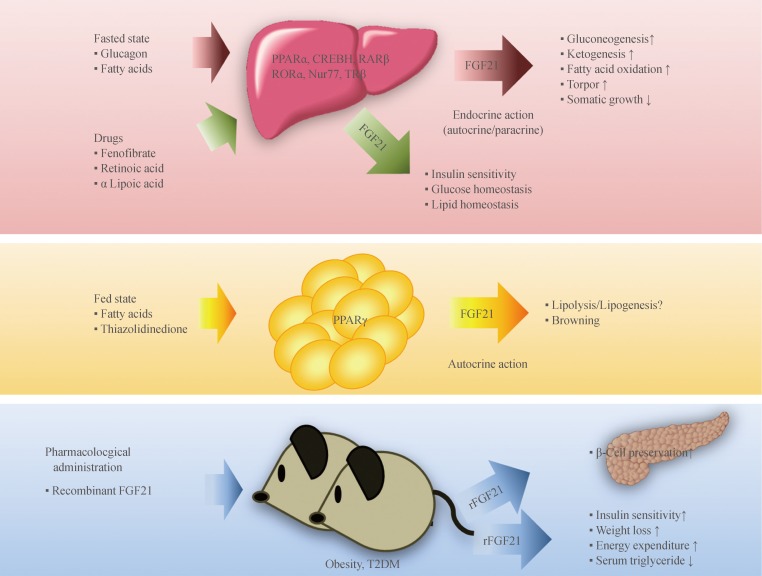
Endocrine, autocrine, and pharmacological actions of fibroblast growth factor 21 (FGF21) and its transcription factors. Fasting induces FGF21 expression in the liver through several transcription factors, and FGF21 acts as an endocrine hormone to induce ketogenesis, gluconeogenesis, fatty acid oxidation, and torpor and to inhibit somatic growth. In the fed state, FGF21 expression in white adipose tissue is induced by peroxisome proliferator-activated receptor gamma (PPARγ), and FGF21 acts through an autocrine or paracrine mechanism to increase PPARγ activity. Pharmacological administration of recombinant FGF21 (rFGF21) affects multiple tissues and has beneficial effects on lipid and glucose metabolism in metabolic disease, including obesity and diabetes mellitus. Recent studies have demonstrated that several metabolically-active drugs produce hepatic FGF21, suggesting a relationship between their actions in glucose and lipid metabolism with the up-regulation of FGF21 production. CREBH, cyclic adenosine monophosphate (AMP) response element-binding protein H; RARβ, retinoic acid (RA) receptor β; RORα, RA receptor-related orphan receptor α; Nur77, nerve growth factor IB; TRβ, thyroid hormone receptor β; T2DM, type 2 diabetes mellitus.
FGF21 AND DIABETES MELLITUS
Serum FGF21 levels and hepatic FGF21 expression increase during obesity or type 2 diabetes [23]. However, although endogenous FGF21 fails to improve glucose and lipid metabolism during obesity [24], pharmacological administration of FGF21 improves not only insulin sensitivity but also β-cell function, which contributes to the beneficial glycemic actions of FGF21 [25]. FGF21 treatment increases islet cell number and insulin staining in db/db mice, demonstrating the ability of FGF21 to preserve β-cell mass and function [25]. Several lines of evidence suggest that FGF21 protects pancreatic β-cells by reducing β-cell glucolipotoxicity and directly reducing β-cell apoptosis via the Akt pathway [25,26,27]. As progressive β-cell loss is important for the pathophysiology of type 2 diabetes, these data suggest that FGF21 could be used to prevent the progression of type 2 diabetes.
TRANSCRIPTIONAL REGULATION OF FGF21 VIA UPREGULATION OF PPARα OR PPARγ
In 2007, three groups found that fasting induces hepatic FGF21 expression and that PPARα is required for this normal starvation response [15,16,28]. PPARα, a nuclear receptor highly expressed in liver, binds directly to the FGF21 gene promoter to induce its transcription [16]. PPARα activation promotes fatty acid oxidation, ketogenesis, and gluconeogenesis [12,29]. PPARα knockout mice accumulate hepatic triglycerides and become hypoketonemic and hypoglycemic during fasting [30]. Hepatocytes are the primary source of circulating FGF21, and its synthesis is driven by the action of PPARα [16]. PPARγ is a nuclear receptor that regulates many genes involved in adipocyte differentiation, lipid synthesis and storage, insulin signaling, and glucose metabolism [31,32]. Activation of PPARγ also increases the secretion of adipokines implicated in whole-body insulin sensitization [33]. Recent studies demonstrated that PPARγ activation increases FGF21 production in adipose tissue, the secondary source of FGF21, which then acts as an autocrine or endocrine factor to improve insulin action [21,34].
REGULATION OF FGF21 EXPRESSION BY OTHER TRANSCRIPTION FACTORS
In addition to PPARα and PPARγ, several studies have suggested that other transcription factors are involved in the regulation of hepatic FGF21 expression. Adams et al. [35]. report that thyroid hormone receptor β, which mediates the action of tri-iodothyronine in the liver, stimulates lipolysis, and hepatic fatty acid oxidation via FGF21 induction. The beneficial metabolic effects of all-trans retinoic acid (RA), which is mediated by RA receptor β (RARβ) binding, are similar to those induced by FGF21, including body weight loss and improvements in glucose and lipid metabolism [36,37]. Several researchers have speculated that FGF21 expression might be regulated by RARβ. FGF21 was characterized as a novel target gene of RARβ in hepatocytes, and hepatic RARβ can bind to putative RA responsive elements in the FGF21 promoter in a fasting-induced manner [38]. RA receptor-related orphan receptor α (RORα) is a nuclear receptor that plays a critical role in lipid metabolism [39], possibly by modulation of FGF21 secretion [40]. A recent study demonstrated that RORα directly regulates the expression and secretion of FGF21 [40]. Cyclic AMP response element-binding protein H (CREBH), an endoplasmic reticulum membrane-bound transcription factor, also induces hepatic FGF21 expression [41,42]. CREBH-deficient mice exhibit impaired fasting-induced expression of FGF21 [43]. Furthermore, CREBH is induced by fenofibrate, which is a well-known PPARα activator. We reported that CREBH mediates fenofibrate-induced suppression of hepatic lipogenesis [44]. Nur77, also known as nuclear receptor subfamily 4 group A member 1 (NR4A1), is a transcription factor in the Nur nuclear hormone receptor superfamily [45]. Nur77 expression is highly induced in adipose tissue, skeletal muscle, and liver by diverse stimuli including β-adrenergic agonists, cold exposure, and fatty acids [46,47,48,49,50,51]. Hepatic Nur77 expression is potently induced by glucagon secretion and fasting, and Nur77 is implicated in hepatic gluconeogenesis [52]. Recently, we found that during fasting, Nur77 mediates hepatic FGF21 expression and that alpha lipoic acid (ALA) increases hepatic FGF21 expression via upregulation of Nur77 (unpublished data).
REGULATION OF FGF21 TRANSCRIPTION BY THERAPEUTIC AGENTS
Increased serum FGF21 concentrations are associated with obesity and insulin resistance in rodents that respond poorly to endogenous FGF21, indicating that metabolic syndrome could be the result of an FGF21-resistant state [22,24]. However, pharmacological administration of FGF21 improves glucose and lipid metabolism in diabetic rhesus monkeys, which present with the same characteristics as diabetic humans [11]. Therefore, regulators of FGF21 transcription, especially in obesity, may be potential drug targets useful for reducing FGF21 resistance. Fenofibrate, a PPARα agonist, is used clinically for the treatment of hypertriglyceridemia [53] and increases FGF21 expression via PPARα activation [54]. As mentioned previously, FGF21 is a downstream target of PPARγ, and the therapeutic effects of PPARα agonists may be mediated by stimulation of hepatic FGF21 production [16]. Thiazolidinediones, a class of antidiabetic drugs (insulin sensitizers), are well-known PPARγ agonists, and FGF21 expression can be regulated by PPARγ agonists in adipose tissue [55]. Cotreatment with FGF21 and a PPARγ agonist results in synergistic adipocyte differentiation and glucose uptake in adipose tissue [19]. Moreover, thiazolidinedione increases FGF21-induced tyrosine phosphorylation of the FGF receptor and induces β-Klotho expression [56,57]. ALA, a naturally occurring thiol antioxidant, is an essential cofactor for mitochondrial respiration [58] and is often used to manage diabetic complications [59,60]. ALA mediates a diverse range of activities including regulation of glucose and lipid metabolism by modulation of PPAR-regulated genes and key enzymes [61]. We previously demonstrated that ALA activates adenosine monophosphate-activated protein kinase and reduces lipid accumulation in livers of rodents fed a high-fat diet [62]. We also reported that ALA enhances Nur77 expression in vascular cells [63] suggesting that ALA treatment may induce nutritionally regulated gene expression in the liver through the upregulation of fasting-induced transcription factors. Recently, we found that ALA increases hepatic FGF21 expression via upregulation of Nur77 and CREBH (unpublished data). Despite many studies on FGF21, its mechanism of action remains controversial and is not fully understood. It is now clear that the FGF21 transcription factor pathway is one of the most promising drug targets for the treatment of metabolic syndrome. Therefore, further studies focused on the transcriptional regulation of FGF21 are necessary.
CONCLUSIONS
FGF21 has emerged as an important hormonal regulator of glucose and lipid metabolism and a promising agent for the treatment of obesity and type 2 diabetes. Therefore, it is necessary to understand the mechanisms responsible for FGF21 expression and identify other transcription factors including nuclear receptors which are able to regulate hepatic FGF21 expression.
ACKNOWLEDGMENTS
This work was supported by grants from the National Research Foundation of Korea (no. 2012R1A2A2A01043867) funded by the Ministry of Science, ICT & Future Planning.
Notes
No potential conflict of interest relevant to this article was reported.