Articles
- Page Path
- HOME > Endocrinol Metab > Volume 36(5); 2021 > Article
-
Original ArticleThyroid Lobeglitazone, A Peroxisome Proliferator-Activated Receptor-Gamma Agonist, Inhibits Papillary Thyroid Cancer Cell Migration and Invasion by Suppressing p38 MAPK Signaling Pathway
-
Jun-Qing Jin
 , Jeong-Sun Han, Jeonghoon Ha, Han-Sang Baek, Dong-Jun Lim
, Jeong-Sun Han, Jeonghoon Ha, Han-Sang Baek, Dong-Jun Lim -
Endocrinology and Metabolism 2021;36(5):1095-1110.
DOI: https://doi.org/10.3803/EnM.2021.1155
Published online: October 14, 2021
Division of Endocrinology and Metabolism, Department of Internal Medicine, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea
- Corresponding author: Dong-Jun Lim. Division of Endocrinology and Metabolism, Department of Internal Medicine, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, Korea, Tel: +82-2-2258-6009, Fax: +82-2-599-3589, E-mail: ldj6026@catholic.ac.kr
Copyright © 2021 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Peroxisome proliferator-activated receptor-gamma (PPAR-γ) ligands have been widely shown to correlate with epithelial-mesenchymal transition (EMT) and cancer progression. Lobeglitazone (LGZ) is a novel ligand of PPAR-γ; and its role in EMT and metastasis in papillary thyroid carcinoma (PTC) is poorly understood. We aimed to investigate the role of LGZ in metastatic behavior of PTC cells.
-
Methods
- Half maximal inhibitory concentration (IC50) values of LGZ in BRAF-mutated PTC cell lines (BCPAP and K1) were determined using MTT assay. Rosiglitazone (RGZ), the PPAR-γ ligand was used as a positive control. The protein expression of PPAR-γ, cell-surface proteins (E-cadherin, N-cadherin), cytoskeletal protein (Vimentin), transcription factor (Snail), p38 mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK) 1/2 pathway, and matrix metalloproteinase (MMP)-2 expression were measured using Western blotting. Changes in E-cadherin expression were also determined using immunocytochemistry. Cell migration and invasion were analyzed using wound healing and Matrigel invasion assays.
-
Results
- Treatment with LGZ or RGZ significantly inhibited transforming growth factor-beta1 (TGF-β1)-induced EMT-associated processes such as fibroblast-like morphological changes, EMT-related protein expression, and increased cell migration and invasion in BCPAP and K1 cells. LGZ restored TGF-β1-induced loss of E-cadherin, as observed using immunocytochemistry. Furthermore, LGZ and RGZ suppressed TGF-β1-induced MMP-2 expression and phosphorylation of p38 MAPK, but not ERK1/2. Although there was no change in PPAR-γ expression after treatment with LGZ or RGZ, the effect of downstream processes mediated by LGZ was hampered by GW9662, a PPAR-γ antagonist.
-
Conclusion
- LGZ inhibits TGF-β1-induced EMT, migration, and invasion through the p38 MAPK signaling pathway in a PPAR-γ-dependent manner in PTC cells.
- Papillary thyroid carcinoma (PTC) is the most common endocrine malignancy and accounts for nearly 90% of all thyroid cancers [1,2]. The rising incidence rate of PTC may be a result of either an increase in the occurrence of this disease or a continuous improvement of its diagnosis [3]. PTC frequently shows local lymph node metastasis or rarely distant metastasis to the lung or bone [4]. Most PTC patients have a good prognosis, with aggressive surgical resection and radioiodine (RI) remnant ablation; however, these effective managements are limited to resectable or RI-avid metastatic PTC [5]. Therefore, there is an unmet need for new therapeutic drugs for RI-refractory thyroid cancers.
- Epithelial-mesenchymal transition (EMT) is a process during which epithelial cells lose their original polarity and are transformed to a mesenchymal phenotype [6,7]. During EMT, cancer cells acquire potent growth signals to obtain invasive capacity, breakdown the basement membrane, and activate the largest process of metastasis [8]. EMT is also defined by the loss of cell-cell adhesion, regulation of cell-surface proteins such as E-cadherin and N-cadherin, induction of cytoskeletal proteins such as Vimentin, and increase the expression of transcription factors such as Snail [9]. Transforming growth factor-beta1 (TGF-β1) is a cytokine belonging to the transforming growth factor superfamily, which has multiple functions and plays a leading role in inducing EMT during cancer metastasis [10].
- TGF-β1 was overexpressed in PTC compared to normal thyroid tissue and high level of TGF-β1 mRNA was detected in the serum of patients with PTC compared with in healthy controls [11]. TGF-β1 plays a major role in promoting tumor invasion during PTC progression [12]. Thyroid cancer from BRAFV600E transgenic mice was susceptible to TGF-β1 induced EMT [13]. In terms of signaling pathways, non-Smad pathways such as mitogen-activated protein kinase (MAPK), Rho, and phosphoinositide 3-kinase (PI3K) signaling, as well as the conventional Smad pathway, were also involved in TGF-β1-induced EMT [14]. Previous studies have demonstrated that TGF-β1 mediated tumor cell EMT and metastasis through the MAPK pathway [15]. Furthermore, tumor cells can migrate and invade target organs by expressing matrix metalloproteinases (MMPs), such as MMP-2, to degrade the extracellular matrix (ECM) proteins [16]. These studies may reveal that clarifying the relationship between EMT and MAPK signaling pathways may play a crucial role in PTC metastasis.
- Increasing evidences of epidemiological, and clinical studies have suggested a link between diabetes and cancer, since patients with diabetes have greater risk of thyroid malignancy [17]. Multiple risk factors including hyperinsulinemia, hyperlipidemia, hyperglycemia, and inflammation potentially contribute to progression of cancer in type 2 diabetes [18]. Earlier clinical study demonstrated that one antidiabetic drug, rosiglitazone (RGZ) used in patients reduce the risk of thyroid cancer [19].
- Previous studies have shown that peroxisome proliferator-activated receptor gamma (PPAR-γ) ligands, such as RGZ, troglitazone, and pioglitazone can inhibit cancer cell proliferation and metastasis in lung, breast, and pancreatic cancers both in vitro and in vivo [20–22]. One earlier clinical study proposed the possible antitumor effects of pioglitazone against metastatic paired box 8 (PAX8)-PPAR-γ fusion protein thyroid carcinoma [23]. Early animal study showed that RGZ effectively inhibit the progression of thyroid carcinogenesis in mouse model [24]. Moreover, it was shown that RGZ induced a partial reversion of EMT in anaplastic thyroid cancer (ATC) cells by multiple mechanisms [25]. All these studies suggest that PPAR-γ agonists have a potential antitumor role in thyroid cancer treatment.
- Lobeglitazone (LGZ), a novel activator of PPAR-γ, is a widely used thiazolidinedione (TZD) for the treatment of type 2 diabetes in Korea [26]. Compared to other TZDs (RGZ and troglitazone), LGZ had an improved safety profile and did not contribute to bladder cancer tumorgenesis [27,28]. However, the anti-tumor effects of LGZ in thyroid cancer have not yet been recognized. In this study, we aimed to show that a new PPAR-γ ligand, LGZ, can prevent TGF-β1-induced cancer progression and metastasis in the most prevalent thyroid cancer, BRAF-mutated PTC cells.
INTRODUCTION
- Cell culture and reagents
- PTC cell lines (BCPAP and K1 were both harboring the BRAFV600E mutation) were grown in Roswell Park Memorial Institute 1640 (RPMI1640) or Dulbecco’s Modified Eagle Medium (DMEM)/F12 (1:1) medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and both were supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2. Recombinant human TGF-β1 (7754-BH, R&D systems, Minneapolis, MN, USA) was reconstituted at 100 μg/mL in sterile 4 mM HCl containing at least 0.1% bovine serum albumin (BSA). LGZ was provided by Chong Kun Dang Pharm. Inc, and another conventional PPAR-γ ligand, RGZ (122320-73-4, Sigma-Aldrich, St. Louis, MO, USA), and PPAR-γ antagonist-2-chloro-5-nitrobenzanilide (GW9662) (M6191, Sigma-Aldrich) were purchased and dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. This study followed Institutional Review Board of Seoul St. Mary’s Hospital, The Catholic University of Korea, which indicated for exemption (Number: KC-20EISI0390).
- Cellular viability assay
- The viability of PTC cells was analyzed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, BCPAP and K1 cells were seeded into a 96-well cell culture plate at a density of 3×103 cells/well in 100 μL of growth media. Following 24 hours incubation at 37°C, cells were treated with LGZ (0.01 to 1,000 μM) or vehicle (DMSO) control and cultured at 37°C for 24 or 48 hours. MTT (298-93-1, ALL for LAB, Seoul, Korea), a stock solution of 5 mg/mL was added to each well and incubated at 37°C for 4 hours. The supernatant was removed. Subsequently, in order to dissolve the formazan crystals, 150 μL of DMSO was added to each well. The plate was shaken for 15 minutes at room temperature (RT). The absorbance was read at 540 nm using an ELISA reader (BioTek, Winooski, VT, USA).
- Western bolt
- Cells were washed twice with phosphate buffered saline (PBS) at 4°C and lysed in RIPA buffer. Protein levels in the cell lysates were measured using a standard Bradford assay. Total protein amount (10 μg) from each sample was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with BSA in Tris-buffered saline, 0.1% Tween® 20 Detergent (TBST) for 2 hours at RT, the membranes were incubated overnight at 4°C with specific primary antibodies: PPAR-γ (1:1,000; sc-7273; Santa Cruz Biotechnology, Dallas, TX, USA), CCAAT/enhancer binding protein alpha (C/EBPα; 1:500; D56F10, Cell Signaling Technology, Danvers, MA, USA), E-cadherin (1:1,000; ab40772, Abcam, Cambridge, UK), N-cadherin (1:1,000; 13116S, Cell Signaling Technology), Vimentin (1:1,000; 5741S, Cell Signaling Technology), Snail (1:1,000; Novus, NBP2-27293, Littleton, CO, USA), p-P38 (1:1,000; 4511, Novus), total-P38 (1:1,000; 9212, Novus), MMP-2 (1:1,000; GTX-104577, Genetex, Irvine, CA, USA), and β-actin (1:1,000; ab8227, Abcam). The membranes were then washed and incubated with the secondary antibodies goat anti-rabbit immunoglobulin G (IgG) or goat anti-mouse IgG (1:2,000; Cell Signaling Technology) for 2 hours with BSA in TBST. The bands were detected using an image analyzer system (Syngene, Cambridge, UK).
- Immunocytochemistry
- BCPAP and K1 cells were trypsinized and seeded on coverslips at 70% confluence. Cells were washed twice with PBS and fixed in 4% formaldehyde in PBS for 10 minutes at RT. The cells were permeabilized with 0.1% TritonX-100 for 60 minutes at RT. The cells were washed three times with PBS. Subsequently, the cells were blocked with 5% normal goat serum in PBS for 3 hours at RT. Fixed cells were incubated with the primary antibody E-cadherin overnight at 4°C. After washing with PBS three times, cells were incubated with secondary antibody Alexa Fluor 488 rabbit anti-rabbit IgG (A-21206, 1:500, Invitrogen) for 45 minutes at RT protected from light. Finally, the cells were washed with PBS three times and washed once with distilled water. The coverslips were mounted with a mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; AR-6501-01, ImmunoBioscience Corp., Mukilteo, WA, USA). Images were captured using a confocal laser scanning microscope LMS800 (Carl Zeiss AG, Oberkochen, Germany) at 640× magnification.
- Cell migration assay
- Cell suspensions containing 4×105 cells/mL were seeded on a 6-well plate. After 24 hours of incubation, the cells in each well reached 100% confluence, and a wound was made by using a sterile 200-μL pipette tip on the cell monolayer. Cells were washed with PBS until there were no floating cells and then serum-free medium was added. Cells were pretreated with the indicated concentrations of RGZ and LGZ for 30 minutes and then stimulated with TGF-β1 (20 ng/mL) for 24 hours. Images were captured using a Nikon Nikon-TS100 microscope (Nikon, Tokyo, Japan). The percentage scratch closure was calculated using ImageJ software.
- Matrigel invasion assay
- Matrigel invasion assays were performed according to the protocol of manufacturer’s instructions (Corning, Acton, MA, USA). A 100-μL of cell suspension (4×104 cells) in serum-free media was added to the upper chamber, while the lower chamber was filled with RPMI or DMEM containing 10% FBS. Each upper and lower chamber was separated by an 8-μm porous polycarbonate membrane. Cells were pretreated with LGZ and RGZ for 15 minutes in the upper chamber and then stimulated by the presence or absence of TGF-β1 (20 ng/mL) for 24 hours and incubated at 37°C with 5% CO2. After the medium was discarded, the cells were washed three times with PBS. Cells at the upper surface of the membrane were removed using a cotton swab. The invaded cells on the bottom of the membrane were fixed with 4% formaldehyde for 30 minutes and stained with hematoxylin and eosin (H&E) protected from light for 20 minutes. For each filter, the number of cells at the lower surface membrane which were counted in five randomly selected areas at 400× magnification under an Axioimager M1 fluorescence microscope (Carl Zeiss). The number of invaded cells was counted using ImageJ software. The mean was calculated from the data obtained from each experiment, and each experiment was repeated in three independent runs.
- Statistical analysis
- All data from at least three independent experiments were normalized to the control. Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). The significance between the two groups was determined using Student’s t test. P<0.01 was considered statistically significant. Error bars represent the mean±standard error of the mean. The half maximal inhibitory concentration (IC50) values and dose response curves were calculated and described using GraphPad Prism 8.0.
METHODS
- Identification of LGZ as a selective PPAR-γ agonist and detecting PPAR-γ expression in BCPAP and K1
- To understand the characteristics of the new PPAR-γ ligand, LGZ, we compared it with the classical TZD, RGZ. The IC50 value of LGZ in PTC has not been previously studied. Through cellular drug toxicity experiments, IC50 value of LGZ in BCPAP cell-line was established as 75.65 μM (24 hours) and 65.32 μM (48 hours), and that in K1 cell-lines was 402.6 μM (24 hours) and 249.0 μM (48 hours), suggesting that BCPAP is more susceptible to LGZ than K1 (Fig. 1A). The PPAR-γ protein expression in BCPAP and K1 was measured using western blotting, and 3T3-L1 cells were used as a positive control (Fig. 1B). BCPAP and K1 cells were treated with RGZ or LGZ for 24 hours. Our results showed that BCPAP and K1 had a very low protein level of PPAR-γ, and no significant difference in PPAR-γ level was observed between RGZ and LGZ treatment compared with the control. As PPAR-γ expression was not definite in BCPAP and K1, expression of C/EBPα, one major protein of the PPAR-γ target genes, was evaluated as a surrogate for PPAR-γ molecular pathway. Western blot result showed that compared with control, C/EBPα expression was increased after RGZ or LGZ treatment, theoretically just as PPAR-γ expression, and this increased expression was attenuated after co-treatment with TGF-β1, but was not attenuated under LGZ co-treatment in BCPAP cells. In K1 cells, on the other hand, C/EBPα expression was increased after RGZ or LGZ treatment (more in LGZ) and decreased after co-treatment with TGF-β1 in LGZ but did not decrease in RGZ, even more increased (Fig. 1C).
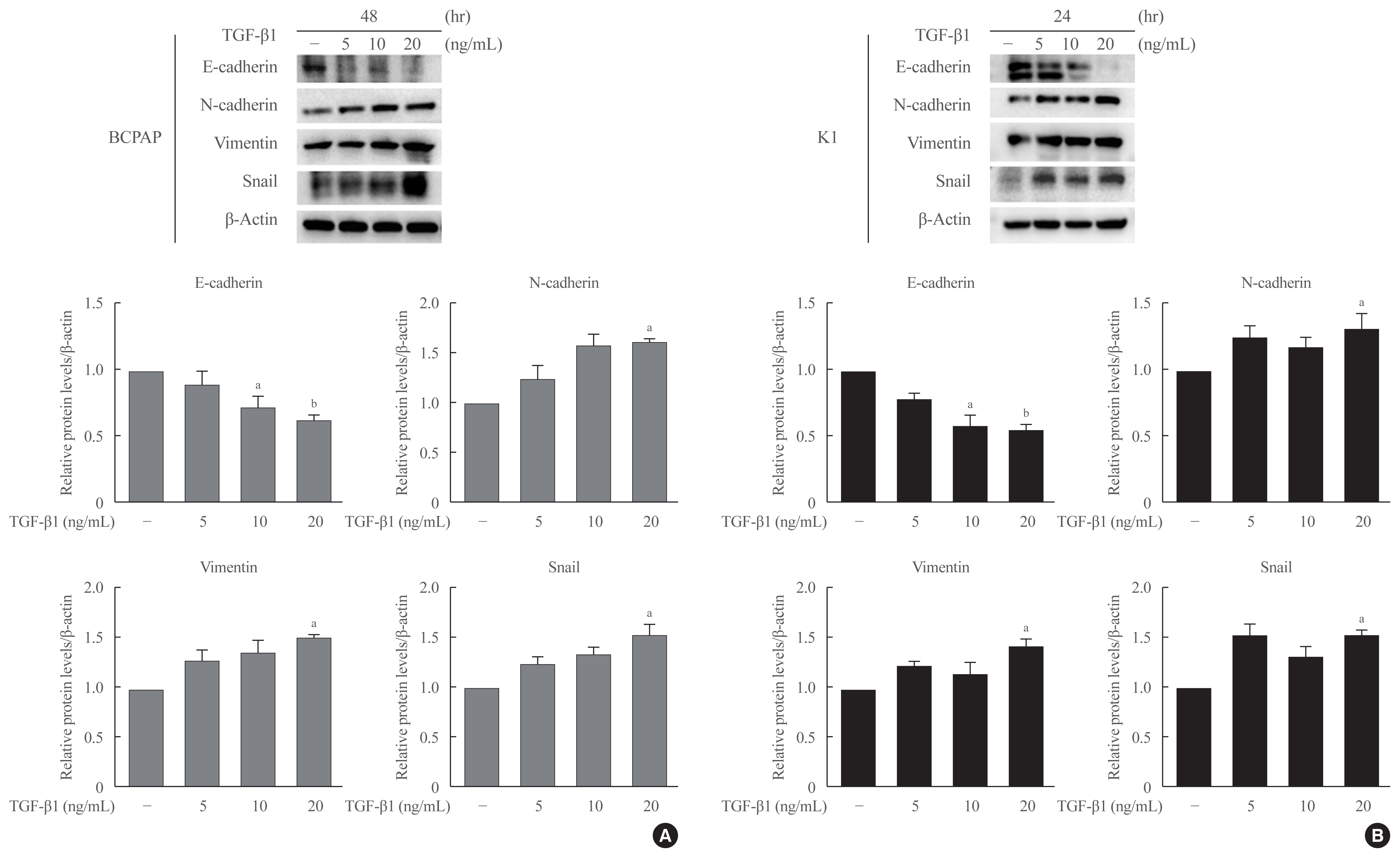
- TGF-β1 induces epithelial-mesenchymal transition in PTC cells
- To determine the optimal concentration and time point at which TGF-β1 induces EMT in PTC cells, we used multiple time points (24, 48, and 72 hours) and concentrations (5, 10, and 20 ng/mL) of TGF-β1 to induce EMT in both PTC cells, BCPAP and K1, and evaluated the results using Western blotting. The expression of E-cadherin was significantly decreased, and the expression of N-cadherin, Vimentin, and Snail was increased by the treatment with TGF-β1 at 20 ng/mL for 48 hours in BCPAP and for 24 hours in K1 (Fig. 2).
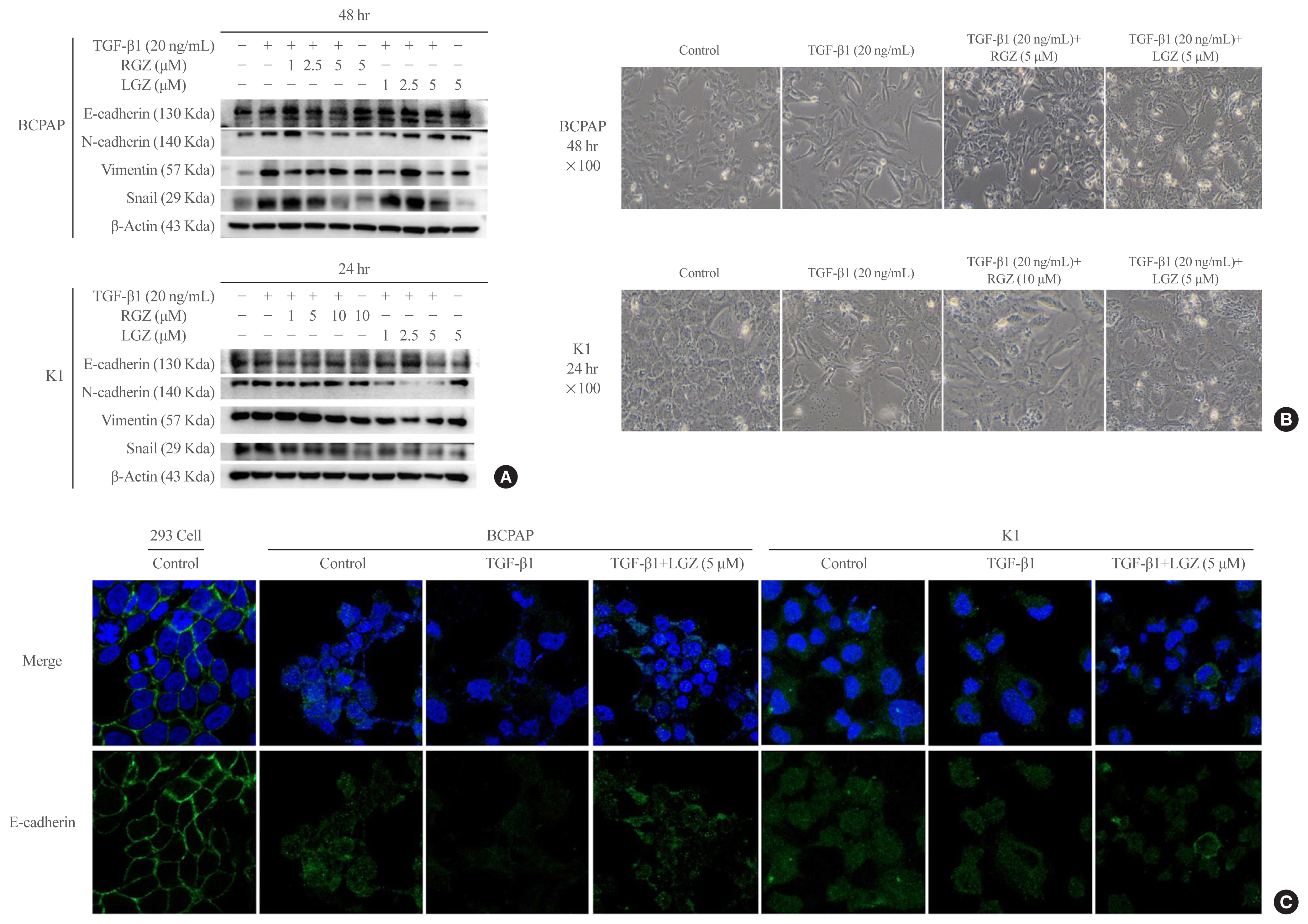
- LGZ and RGZ inhibit TGF-β1-induced EMT in PTC cells
- We used western blotting to investigate the effect of PPAR-γ ligands RGZ or LGZ on TGF-β1-induced EMT in PTC cells. The results showed that the expression of E-cadherin was increased and the expression of N-cadherin, Vimentin, and Snail was decreased in a dose-depended manner in response to RGZ and LGZ treatment compared with TGF-β1 treatment alone at 24- and 48-hour time points in PTC cells (Fig. 3A). Stimulation with TGF-β1 (20 ng/mL) in the presence or absence of RGZ (5 μM) or LGZ (5 μM) at 48 hours in BCPAP and RGZ (10 μM) or LGZ (5 μM) at 24 hours in K1 showed that the typical cellular morphological changes corresponding to the EMT phenotype, including loss of cell-to-cell junctions, and increased intercellular space, were induced and reversed by RGZ and LGZ. In contrast, PPAR-γ ligands completely prevented the cells from undergoing the EMT morphological changes induced by TGF-β1 (Fig. 3B). Based on these results, we used 5 μM LGZ (BCPAP and K1) and 5 μM RGZ (BCPAP) and 10 μM RGZ (K1) to maximally inhibit the TGF-β1-induced EMT in the following experiments. Immunocytochemical staining confirmed that treating with LGZ 5μM recovered E-cadherin expression, which was reduced by TGF-β1 in both BCPAP and K1 cells (Fig. 3C).
- LGZ and RGZ inhibit TGF-β1-induced EMT in PTC cells through the p38 MAPK signaling pathway
- Our experiments showed that p38 MAPK phosphorylation was activated by TGF-β1 in a time-dependent manner. p38 phosphorylation was significantly increased by TGF-β1 in a time-dependent manner (Fig. 4A). RGZ and LGZ decreased the level of p38 MAPK phosphorylation induced by TGF-β1 after 15 minutes. In contrast, total p38 levels were unaffected by the addition of TGF-β1, RGZ, and LGZ (Fig. 4B). We also found that LGZ and RGZ did not have a significant effect on extracellular signal-regulated kinase (ERK) 1/2 phosphorylation induced by TGF-β1 in BCPAP and K1 cells (Fig. 4C).
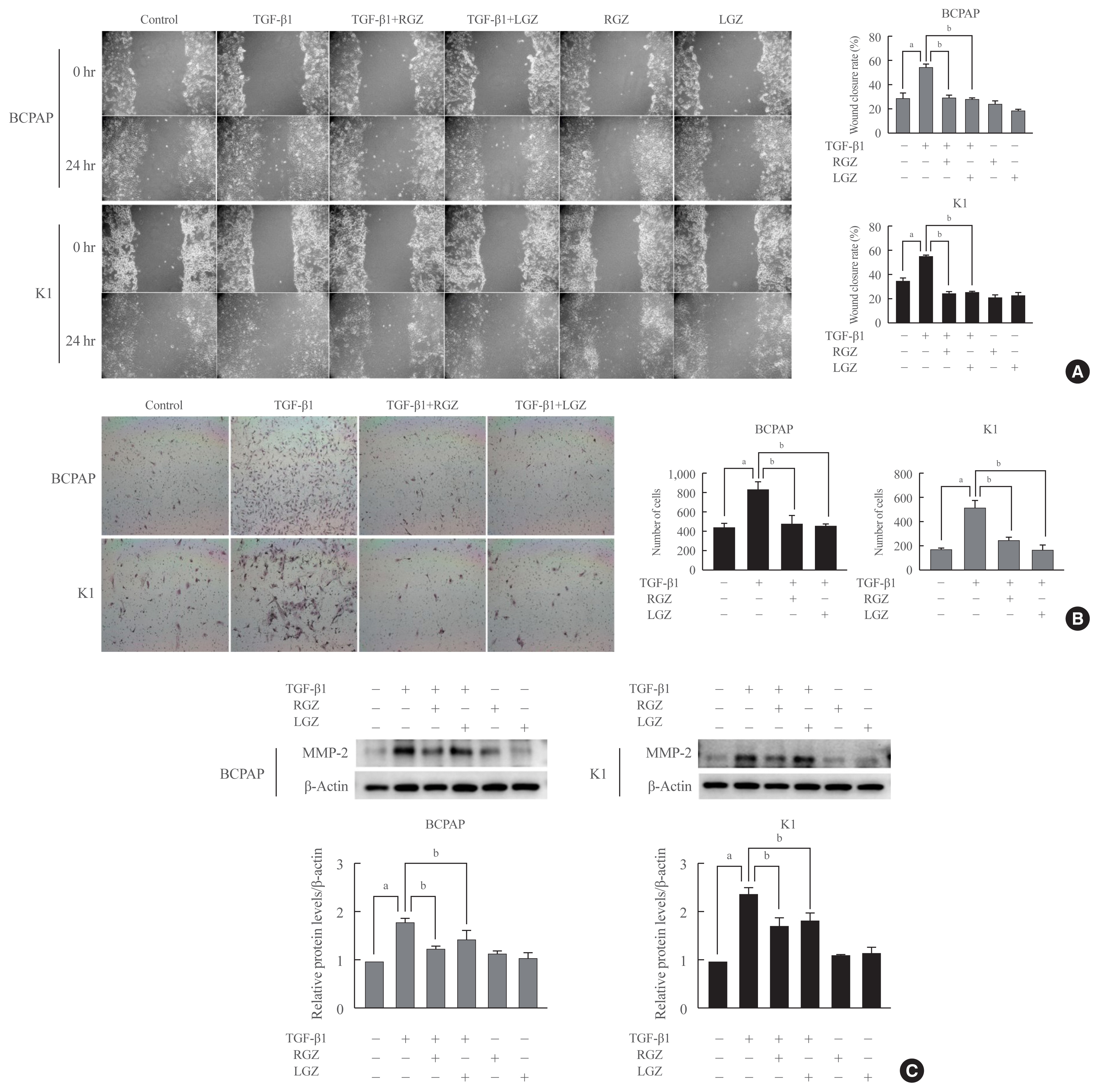
- LGZ and RGZ inhibit cell migration, invasion, and MMP-2 protein expression induced by TGF-β1 in PTC cells
- To assess the effect of PPAR-γ ligands on TGF-β1-induced PTC migration and invasion, we performed wound healing and Matrigel invasion assays. BCPAP and K1 were pretreated with RGZ and LGZ for 30 min and then treated with TGF-β1 for 24 hours. As the results showed, RGZ and LGZ significantly inhibited TGF-β1-induced cell migration in PTC cells (Fig. 5A). However, RGZ or LGZ stimulation alone did not affect PTC cell migration. In Matrigel invasion, similar to the migration assay results, RGZ and LGZ significantly reduced TGF-β1-induced invasion (Fig. 5B). Furthermore, we assessed MMP-2 expression in PTC cells using western blotting. In both BCPAP and K1 cells, baseline MMP-2 expression was not observed but could be induced by TGF-β1. RGZ and LGZ significantly inhibited TGF-β1-induced MMP-2 expression (Fig. 5C). These results suggest that RGZ and LGZ can inhibit the migration and invasion of thyroid cancer cells in the EMT induced by TGF-β1.
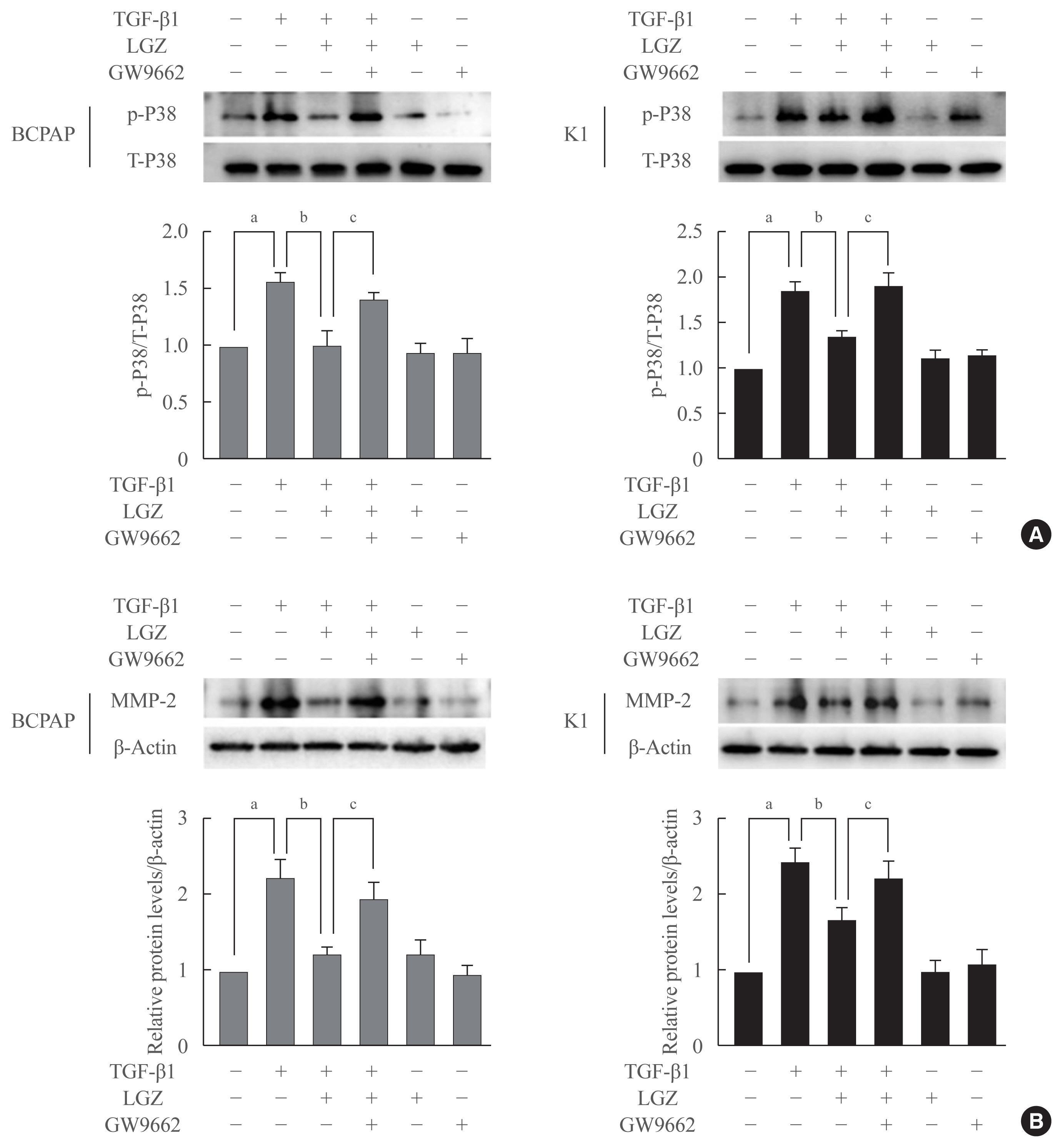
- Antitumor effects of LGZ are PPAR-γ dependent
- To further assess whether the effects of LGZ on phosphorylation of p38 and MMP-2 expression were PPAR-γ dependent, we used the PPAR-γ selective antagonist GW9662 to block PPAR-γ activity. BCPAP and K1 cells were pretreated with GW9662 for 2 hours, and then stimulated with TGF-β1. GW9662 restored LGZ-induced downregulation of p38 and MMP-2 expression in both BCPAP and K1 cells (Fig. 6), indicating that the effects of LGZ on phosphorylation of p38 and MMP-2 secretion are PPAR-γ-dependent.
RESULTS
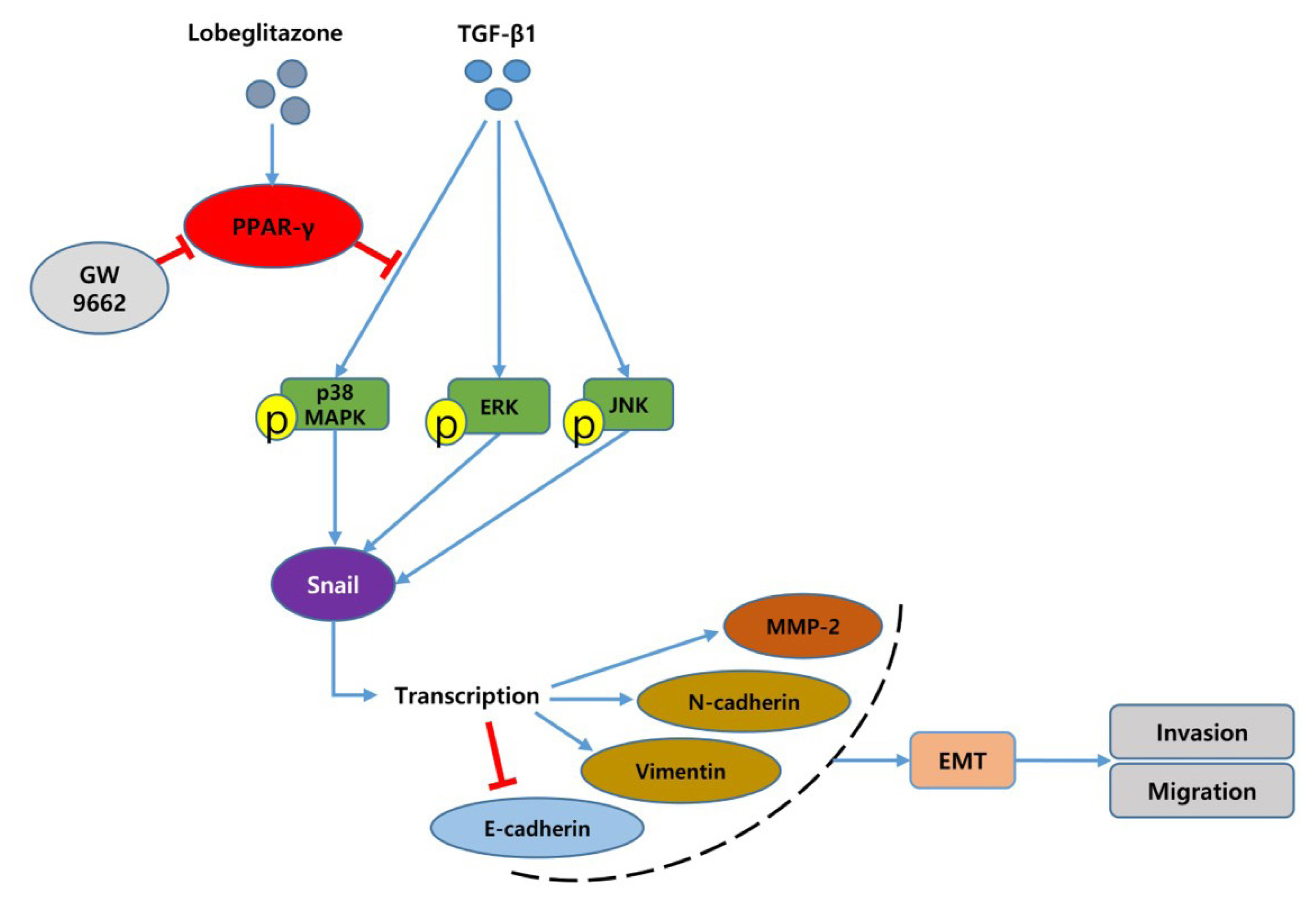
- In this study, we investigated the role of LGZ in metastatic behavior of PTC cells. Activation of PPAR-γ by TZDs can inhibit cellular proliferation, migration, and invasion in many types of cancer cells through a PPAR-γ-dependent or -independent pathway [29,30]. These findings indicate that PPAR-γ ligands have the potential to prevent cancer metastasis. In our study, we showed that LGZ and RGZ could prevent TGF-β1-induced EMT, migration, and invasion in PTC cells. We used different time points to treat TGF-β1 (48 hours in BCPAP and 24 hours in K1) because these conditions indicated the typical EMT phenotypes with peak changes of EMT-related molecular markers. Dosages of LGZ (5 μM) and RGZ (10 μM) were in accordance with the reference experiments of EMT in renal cells (LGZ) [31], lung cancer cells and anaplastic thyroid cancer cells [21,25]. Furthermore, LGZ also downregulated MMP-2 expression and p38 phosphorylation induced by TGF-β1 in a PPAR-γ-dependent manner (Fig. 7). Although, PPAR-γ is known as nuclear receptors, when upon ligand binding, PPAR-γ can exert both nuclear genomic and cytoplasmic non-genomic effects. Cytoplasmic PPAR-γ can also modulate MAPKs signaling pathways and it occurs in a receptor-dependent or receptor-independent manner [32,33].
- It was shown that when PPAR-γ was overexpressed in BCPAP cells lacking PPAR-γ expression, there was increased growth and increased pRb and cyclin A and B1 levels [34]. However, there was no rigorous correlation between the degree of PPAR-γ expression and functional level and biological response to TZDs, such as RGZ, in thyroid cancer cell lines [25]. Similar to these results, we did not observe increased PPAR-γ protein levels in response to the treatment by LGZ or RGZ in BCPAP and K1. Thus, we chose C/EBPα, one major target gene, as a surrogate of PPAR-γ expression, to evaluate the effects of PPAR-γ agonists and TGF-β1 on PPAR-γ molecular pathway.
- Many target genes of PPAR-γ have been already reported, including Fabp4/aP2, C/EBPα, Cfd/Adipsin, CD36 and lipoprotein lipase (LpL) [35–37]. Among them, C/EBPα is a member of subfamily of the basic region leucine zipper transcription factors [38]. It was shown that pioglitazone induced expression of a broad array of PPAR-γ target genes including C/EBPα in mice with paired box gene 8 fusion protein thyroid carcinomas [39]. Previous researches also reported that C/EBPα can inhibit lung cancer development through the p38α MAPK pathway and suppress lung cancer cell invasion and migration [40,41]. As the results showed, C/EBPα expression was increased and decreased to varying degrees by RGZ or LGZ when co-treatment with TGF-β1, maybe due to different mutational profiles of two thyroid cancer cell-lines [42]. Most of all, the relationship between one PPAR-γ target gene, C/EBPα and the EMT-related genes also has been well recognized. Through inhibiting the expression of key mesenchymal markers, C/EBPα prevents EMT-mediated tumorigenesis and maintains epithelial hemostasis [43].
- We showed that the potent, selective, and irreversible PPAR-γ antagonist GW9662 prevented LGZ-mediated downstream processes, including the p38 MAPK pathway and MMP2 expression. The transactivation of the different genes by PPAR-γ activation may vary in different cells according to response elements [44].
- In earlier studies, preventing the depletion of E-cadherin expression blocked the gain of mesenchymal markers and functional phenotypes of motility and invasion during EMT in human breast cancer and hepatoma cancer cells [45,46]. Activation of PPAR-γ can inhibit the loss of E-cadherin induced by TGF-β1, maintaining a differentiated epithelial phenotype in human prostate cancer cell lines [47]. Therefore, preventing the loss of E-cadherin using TZDs may be another important strategy to inhibit EMT. The present study showed that in an in vitro EMT model induced by TGF-β1 treatment, LGZ and RGZ increased E-cadherin expression and decreased N-cadherin, Vimentin, and Snail expression in PTC cells in a dose-dependent manner. Thus, LGZ has clinical potential as an anti-tumor therapy for PTC metastasis.
- Plausible mechanisms underlying the effects of PPAR-γ ligands on TGF-β1 induced EMT, via a Smad or non-Smad pathway, may be cell type-specific and depend on the various types of somatic mutations in cancer cells. Although two PTC cell lines used in our study, BCPAP and K1, have common BRAFV600E mutation, which is the most frequent driver mutation (nearly 80%) in Korea, these two cell types have different mutational profiles [42]: K1 has phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PI3KCA) mutation (E542K) in addition to BRAFV600E mutation so may show different response from BCPAP to TGF-β1 and PPAR-γ agonists, because of constitutive kinase activation. Contrary to expectations, however, BCPAP and K1 showed almost similar results, indicating PPAR-γ agonists might be effective to PTC cells without regard to mutational differences.
- Although TGFβ/Smad signaling has been recognized as the most important pathway in EMT, the non-Smad pathway, including MAPK signaling pathways, might participate in EMT and tumor progression in thyroid cancer [48]. The execution of the EMT program in response to TGF-β depends on the initiating events during tumor microevolution [49]. In BRAFV600E mutated PTC, constitutive activation of the MAPK pathway promotes tumor progression. P38, the major component of the MAPK pathway in the non-SMAD pathway, is phosphorylated by TGF-β1, which binds to its cell surface receptors (TβRI, TβRII) [50,51]. MAPK can activate avian erythroblastosis virus E26 (v-ets) oncogene homolog-1 (ETS-1) and activated ETS-1 cooperates with Snail 1 to upregulate MMP expression [52]. Consistently, we showed that the MAPK-ERK and p38 phosphorylation as well as the increased expression of Snail and MMP-2 were induced by TGF-β1 in a time-dependent manner. Inhibition of MAPK-ERK activity could block EMT process in normal murine mammary gland epithelia cell [53], and activation of MAPK p38 may lead to EMT by TGF-β1 in mammary epithelial cells [54]. It was also demonstrated that MAPK p38-driven MAPK-activated protein kinase 2 regulated the invasive ability of bladder cancer through modulating MMP-2 and MMP-9 activities [55]. Therefore, the MAPK-ERK or p38 signaling pathway might be essential for the EMT process and tumor metastasis induced by TGF-β1 [56]; and inhibition of MAPK or p38 activation could completely suppress the EMT process.
- The effects of RGZ and troglitazone on TGF-β1-induced Smad signaling pathway in lung and pancreatic cancer cells have been well recognized [21]. A previous study demonstrated that pioglitazone had an anti-proliferative effect through the MAPK/AKT cascade as well as the TGFβ/Smad system, thereby exerting anti-metastatic effects by repressing EMT process in human non-small lung cancer cells [57]. Consistent with this observation, both LGZ and RGZ downregulated the MAPK p38 phosphorylation induced by TGF-β1 in our results. Similar to our results, RGZ suppressed fibrotic response by lowering p38 activation but not ERK activation in human pterygium fibroblasts [58]. However, LGZ and RGZ did not suppress the phosphorylation of TGF-β1-induced ERK, another key player in the MAPK pathway. One previous study demonstrated that RGZ inhibited phosphorylation of ERK and Smad2/3 induced by TGF-β1 and no effect on the MAPK p38 in chondrocytes [59]. However, our study suggested that RGZ and LGZ might selectively suppress MAPK p38 to inhibit EMT induced by TGF-β1 in PTC.
- In conclusion, the present study revealed that LGZ abrogated EMT, cellular migration, and invasion induced by TGF-β1 in PTC cells through MAPK p38 signaling. Thus, LGZ has a potential to treat a later-stage PTC with metastasis, and we carefully predict new indication of LGZ above anti-diabetic drug. However, as this study did not show any clinical efficacy of LGZ in in vivo model, it should be evaluated whether a PPAR-γ ligand, LGZ may have a clinical role in suppressing metastatic tumors in animal models by aggressive PTC in the near future.
DISCUSSION
-
Acknowledgements
- This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1-A02045433).
-
CONFLICTS OF INTEREST
This research was supported by the Chong Kun Dang Pharmaceutical Corp., Seoul, Republic of Korea. The sponsor had no role in the writing of the manuscript or in the decision to submit the manuscript for publication.
-
AUTHOR CONTRIBUTIONS
Conception or design: J.Q.J., J.S.H., D.J.L. Acquisition, analysis, or interpretation of data: J.Q.J., J.S.H., J.H., H.S.B., D.J.L. Drafting the work or revising: J.Q.J., D.J.L. Final approval of the manuscript: J.Q.J., J.S.H., J.H., H.S.B., D.J.L.
Article information



- 1. Pellegriti G, Frasca F, Regalbuto C, Squatrito S, Vigneri R. Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J Cancer Epidemiol 2013 2013:965212.Article
- 2. Lubitz CC, Sosa JA. The changing landscape of papillary thyroid cancer: epidemiology, management, and the implications for patients. Cancer 2016;122:3754–9.ArticlePubMed
- 3. Morris LG, Tuttle RM, Davies L. Changing trends in the incidence of thyroid cancer in the United States. JAMA Otolaryngol Head Neck Surg 2016;142:709–11.ArticlePubMedPMC
- 4. LiVolsi VA. Papillary thyroid carcinoma: an update. Mod Pathol 2011;24(Suppl 2):S1–9.ArticlePubMed
- 5. Lebastchi AH, Callender GG. Thyroid cancer. Curr Probl Cancer 2014;38:48–74.ArticlePubMed
- 6. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009;19:156–72.ArticlePubMedPDF
- 7. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871–90.ArticlePubMed
- 8. Wei SC, Fattet L, Yang J. The forces behind EMT and tumor metastasis. Cell Cycle 2015;14:2387–8.ArticlePubMedPMC
- 9. Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers (Basel) 2017;9:171.ArticlePubMedPMC
- 10. Javelaud D, Mauviel A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int J Biochem Cell Biol 2004;36:1161–5.PubMed
- 11. Vasko V, Espinosa AV, Scouten W, He H, Auer H, Liyanarachchi S, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci U S A 2007;104:2803–8.ArticlePubMedPMC
- 12. Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M, et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res 2009;69:8317–25.ArticlePubMed
- 13. Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, et al. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 2011;30:3153–62.ArticlePubMedPMC
- 14. Atfi A, Djelloul S, Chastre E, Davis R, Gespach C. Evidence for a role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor beta-mediated signaling. J Biol Chem 1997;272:1429–32.PubMed
- 15. Fu H, He Y, Qi L, Chen L, Luo Y, Chen L, et al. cPLA2α activates PI3K/AKT and inhibits Smad2/3 during epithelial-mesenchymal transition of hepatocellular carcinoma cells. Cancer Lett 2017;403:260–70.ArticlePubMed
- 16. Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell 1997;91:439–42.ArticlePubMed
- 17. Aschebrook-Kilfoy B, Sabra MM, Brenner A, Moore SC, Ron E, Schatzkin A, et al. Diabetes and thyroid cancer risk in the National Institutes of Health-AARP Diet and Health Study. Thyroid 2011;21:957–63.ArticlePubMedPMC
- 18. Hsu IR, Kim SP, Kabir M, Bergman RN. Metabolic syndrome, hyperinsulinemia, and cancer. Am J Clin Nutr 2007;86:s867–71.ArticlePubMed
- 19. Tseng CH. Rosiglitazone may reduce thyroid cancer risk in patients with type 2 diabetes. Ann Med 2013;45:539–44.ArticlePubMed
- 20. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol 2004;5:419–29.PubMed
- 21. Reka AK, Kurapati H, Narala VR, Bommer G, Chen J, Standiford TJ, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther 2010;9:3221–32.ArticlePubMedPMC
- 22. Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A 1998;95:8806–11.PubMedPMC
- 23. Giordano TJ, Haugen BR, Sherman SI, Shah MH, Caoili EM, Koenig RJ. Pioglitazone therapy of PAX8-PPARγ fusion protein thyroid carcinoma. J Clin Endocrinol Metab 2018;103:1277–81.ArticlePubMedPMC
- 24. Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, et al. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene 2006;25:2736–47.ArticlePubMed
- 25. Aiello A, Pandini G, Frasca F, Conte E, Murabito A, Sacco A, et al. Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology 2006;147:4463–75.PubMed
- 26. Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W, et al. Kinetics of the absorption, distribution, metabolism, and excretion of lobeglitazone, a novel activator of peroxisome proliferator-activated receptor gamma in rats. J Pharm Sci 2015;104:3049–59.ArticlePubMed
- 27. Moon KS, Lee JE, Lee HS, Hwang IC, Kim DH, Park HK, et al. CKD-501, a novel selective PPARγ agonist, shows no carcinogenic potential in ICR mice following oral administration for 104 weeks. J Appl Toxicol 2014;34:1271–84.ArticlePubMed
- 28. Lee HS, Chang M, Lee JE, Kim W, Hwang IC, Kim DH, et al. Carcinogenicity study of CKD-501, a novel dual peroxisome proliferator-activated receptors α and γ agonist, following oral administration to Sprague Dawley rats for 94–101 weeks. Regul Toxicol Pharmacol 2014;69:207–16.ArticlePubMed
- 29. Nemenoff RA. Peroxisome proliferator-activated receptor-gamma in lung cancer: defining specific versus “off-target” effectors. J Thorac Oncol 2007;2:989–92.PubMed
- 30. Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther 2006;5:430–7.ArticlePubMed
- 31. Bae KH, Seo JB, Jung YA, Seo HY, Kang SH, Jeon HJ, et al. Lobeglitazone, a novel peroxisome proliferator-activated receptor γ agonist, attenuates renal fibrosis caused by unilateral ureteral obstruction in mice. Endocrinol Metab (Seoul) 2017;32:115–23.ArticlePubMedPMC
- 32. Vella V, Nicolosi ML, Giuliano S, Bellomo M, Belfiore A, Malaguarnera R. PPAR-γ agonists as antineoplastic agents in cancers with dysregulated IGF axis. Front Endocrinol (Lausanne) 2017;8:31.ArticlePubMedPMC
- 33. Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 2005;70:177–88.ArticlePubMed
- 34. Wood WM, Sharma V, Bauerle KT, Pike LA, Zhou Q, Fretwell DL, et al. PPARγ promotes growth and invasion of thyroid cancer cells. PPAR Res 2011 2011:171765.
- 35. Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994;79:1147–56.ArticlePubMed
- 36. Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A 2009;106:22504–9.PubMedPMC
- 37. Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev 2008;22:2941–52.ArticlePubMedPMC
- 38. Miller M, Shuman JD, Sebastian T, Dauter Z, Johnson PF. Structural basis for DNA recognition by the basic region leucine zipper transcription factor CCAAT/enhancer-binding protein alpha. J Biol Chem 2003;278:15178–84.PubMed
- 39. Dobson ME, Diallo-Krou E, Grachtchouk V, Yu J, Colby LA, Wilkinson JE, et al. Pioglitazone induces a proadipogenic antitumor response in mice with PAX8-PPARgamma fusion protein thyroid carcinoma. Endocrinology 2011;152:4455–65.ArticlePubMedPMC
- 40. Sato A, Yamada N, Ogawa Y, Ikegami M. CCAAT/enhancer-binding protein-α suppresses lung tumor development in mice through the p38α MAP kinase pathway. PLoS One 2013;8:e57013.ArticlePubMedPMC
- 41. Lu J, Du C, Yao J, Wu B, Duan Y, Zhou L, et al. C/EBPα suppresses lung adenocarcinoma cell invasion and migration by inhibiting β-catenin. Cell Physiol Biochem 2017;42:1779–88.ArticlePubMed
- 42. Saiselet M, Floor S, Tarabichi M, Dom G, Hebrant A, van Staveren WC, et al. Thyroid cancer cell lines: an overview. Front Endocrinol (Lausanne) 2012;3:133.ArticlePubMedPMC
- 43. Lourenco AR, Roukens MG, Seinstra D, Frederiks CL, Pals CE, Vervoort SJ, et al. C/EBPα is crucial determinant of epithelial maintenance by preventing epithelial-to-mesenchymal transition. Nat Commun 2020;11:785.ArticlePubMedPMC
- 44. Qi C, Zhu Y, Reddy JK. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem Biophys 2000;32:187–204.ArticlePubMed
- 45. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 2008;68:3645–54.ArticlePubMed
- 46. Lee HJ, Su Y, Yin PH, Lee HC, Chi CW. PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer Res 2009;29:5057–63.PubMed
- 47. Chang SN, Lee JM, Oh H, Kim U, Ryu B, Park JH. Troglitazone inhibits the migration and invasion of PC-3 human prostate cancer cells by upregulating E-cadherin and glutathione peroxidase 3. Oncol Lett 2018;16:5482–8.ArticlePubMedPMC
- 48. Zhou Q, Chen J, Feng J, Xu Y, Zheng W, Wang J. SOSTDC1 inhibits follicular thyroid cancer cell proliferation, migration, and EMT via suppressing PI3K/Akt and MAPK/Erk signaling pathways. Mol Cell Biochem 2017;435:87–95.ArticlePubMed
- 49. Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem 2007;101:816–29.ArticlePubMed
- 50. Palona I, Namba H, Mitsutake N, Starenki D, Podtcheko A, Sedliarou I, et al. BRAFV600E promotes invasiveness of thyroid cancer cells through nuclear factor kappaB activation. Endocrinology 2006;147:5699–707.ArticlePubMed
- 51. Zou M, Baitei EY, BinEssa HA, Al-Mohanna FA, Parhar RS, St-Arnaud R, et al. Cyp24a1 attenuation limits progression of BrafV600E-induced papillary thyroid cancer cells and sensitizes them to BRAFV600E inhibitor PLX4720. Cancer Res 2017;77:2161–72.ArticlePubMedPMC
- 52. Jorda M, Olmeda D, Vinyals A, Valero E, Cubillo E, Llorens A, et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci 2005;118(Pt 15):3371–85.ArticlePubMed
- 53. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004;6:603–10.ArticlePubMedPMC
- 54. Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J 2002;21:3749–59.ArticlePubMedPMC
- 55. Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham RB, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res 2010;70:832–41.ArticlePubMed
- 56. Chapnick DA, Warner L, Bernet J, Rao T, Liu X. Partners in crime: the TGFβ and MAPK pathways in cancer progression. Cell Biosci 2011;1:42.ArticlePubMedPMC
- 57. Ciaramella V, Sasso FC, Di Liello R, Corte CM, Barra G, Viscardi G, et al. Activity and molecular targets of pioglitazone via blockade of proliferation, invasiveness and bioenergetics in human NSCLC. J Exp Clin Cancer Res 2019;38:178.ArticlePubMedPMC
- 58. Nuwormegbe SA, Sohn JH, Kim SW. A PPAR-gamma agonist rosiglitazone suppresses fibrotic response in human pterygium fibroblasts by modulating the p38 MAPK pathway. Invest Ophthalmol Vis Sci 2017;58:5217–26.ArticlePubMed
- 59. Poleni PE, Bianchi A, Etienne S, Koufany M, Sebillaud S, Netter P, et al. Agonists of peroxisome proliferators-activated receptors (PPAR) alpha, beta/delta or gamma reduce transforming growth factor (TGF)-beta-induced proteoglycans’ production in chondrocytes. Osteoarthritis Cartilage 2007;15:493–505.ArticlePubMed
References
Figure & Data
References
Citations
- Diabetes Mellitus and Thyroid Cancers: Risky Correlation, Underlying Mechanisms and Clinical Prevention
Rongqian Wu, Junping Zhang, Guilin Zou, Shanshan Li, Jinying Wang, Xiaoxinlei Li, Jixiong Xu
Diabetes, Metabolic Syndrome and Obesity.2024; Volume 17: 809. CrossRef - Clinicopathological Evaluation of Papillary Thyroid Microcarcinoma
Ando Takahito, Kimihito Fujii, Hirona Banno, Masayuki Saito, Yukie Ito, Mirai Ido, Manami Goto, Yukako Mouri, Junko Kousaka, Tsuneo Imai, Shogo Nakano
Cureus.2024;[Epub] CrossRef - Pioglitazone, a peroxisome proliferator‑activated receptor γ agonist, induces cell death and inhibits the proliferation of hypoxic HepG2 cells by promoting excessive production of reactive oxygen species
Guohao Huang, Mengfan Zhang, Manzhou Wang, Wenze Xu, Xuhua Duan, Xinwei Han, Jianzhuang Ren
Oncology Letters.2024;[Epub] CrossRef - The Activation of PPARγ by (2Z,4E,6E)-2-methoxyocta-2,4,6-trienoic Acid Counteracts the Epithelial–Mesenchymal Transition Process in Skin Carcinogenesis
Enrica Flori, Sarah Mosca, Giorgia Cardinali, Stefania Briganti, Monica Ottaviani, Daniela Kovacs, Isabella Manni, Mauro Truglio, Arianna Mastrofrancesco, Marco Zaccarini, Carlo Cota, Giulia Piaggio, Mauro Picardo
Cells.2023; 12(7): 1007. CrossRef - Cumulative exposure to metabolic syndrome increases thyroid cancer risk in young adults: a population-based cohort study
Jinyoung Kim, Kyungdo Han, Mee Kyoung Kim, Ki-Hyun Baek, Ki-Ho Song, Hyuk-Sang Kwon
The Korean Journal of Internal Medicine.2023; 38(4): 526. CrossRef - Drug repositioning in thyroid cancer treatment: the intriguing case of anti-diabetic drugs
Alessia Greco, Francesca Coperchini, Laura Croce, Flavia Magri, Marsida Teliti, Mario Rotondi
Frontiers in Pharmacology.2023;[Epub] CrossRef - Fish and the Thyroid: A Janus Bifrons Relationship Caused by Pollutants and the Omega-3 Polyunsaturated Fatty Acids
Salvatore Benvenga, Fausto Famà, Laura Giovanna Perdichizzi, Alessandro Antonelli, Gabriela Brenta, Francesco Vermiglio, Mariacarla Moleti
Frontiers in Endocrinology.2022;[Epub] CrossRef - Identifying and categorizing compounds that reduce corneal transforming growth factor beta induced protein levels: a scoping review
Gabriella Guo Sciriha, Janet Sultana, Joseph Borg
Expert Review of Clinical Pharmacology.2022; 15(12): 1423. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite