The Role of Thyroid Hormone in the Regulation of Cerebellar Development
Article information

Abstract
The proper organized expression of specific genes in time and space is responsible for the organogenesis of the central nervous system including the cerebellum. The epigenetic regulation of gene expression is tightly regulated by an intrinsic intracellular genetic program, local stimuli such as synaptic inputs and trophic factors, and peripheral stimuli from outside of the brain including hormones. Some hormone receptors are expressed in the cerebellum. Thyroid hormones (THs), among numerous circulating hormones, are well-known major regulators of cerebellar development. In both rodents and human, hypothyroidism during the postnatal developmental period results in abnormal morphogenesis or altered function. THs bind to the thyroid hormone receptors (TRs) in the nuclei and with the help of transcriptional cofactors regulate the transcription of target genes. Gene regulation by TR induces cell proliferation, migration, and differentiation, which are necessary for brain development and plasticity. Thus, the lack of TH action mediators may directly cause aberrant cerebellar development. Various kinds of animal models have been established in a bid to study the mechanism of TH action in the cerebellum. Interestingly, the phenotypes differ greatly depending on the models. Herein we summarize the actions of TH and TR particularly in the developing cerebellum.
INTRODUCTION
The proper organized expression of specific genes in time and space is responsible for the organogenesis of central nervous system (CNS) [1]. Developmental defects in the brain are induced by abnormal amount, timing, or area of gene expression. The epigenetic regulation of gene expression is tightly regulated by an intrinsic intracellular genetic program in the neuronal cells themselves, and is also regulated by several stimuli; local and peripheral, from other types of cells (Fig. 1). Local stimuli include synaptic inputs and trophic factors within the brain. Peripheral stimuli from outside of the brain include sensory inputs from the peripheral nervous system and hormonal inputs from endocrine cells. These factors all work together contributing to brain development and plasticity. In addition, environmental factors are important modulators.
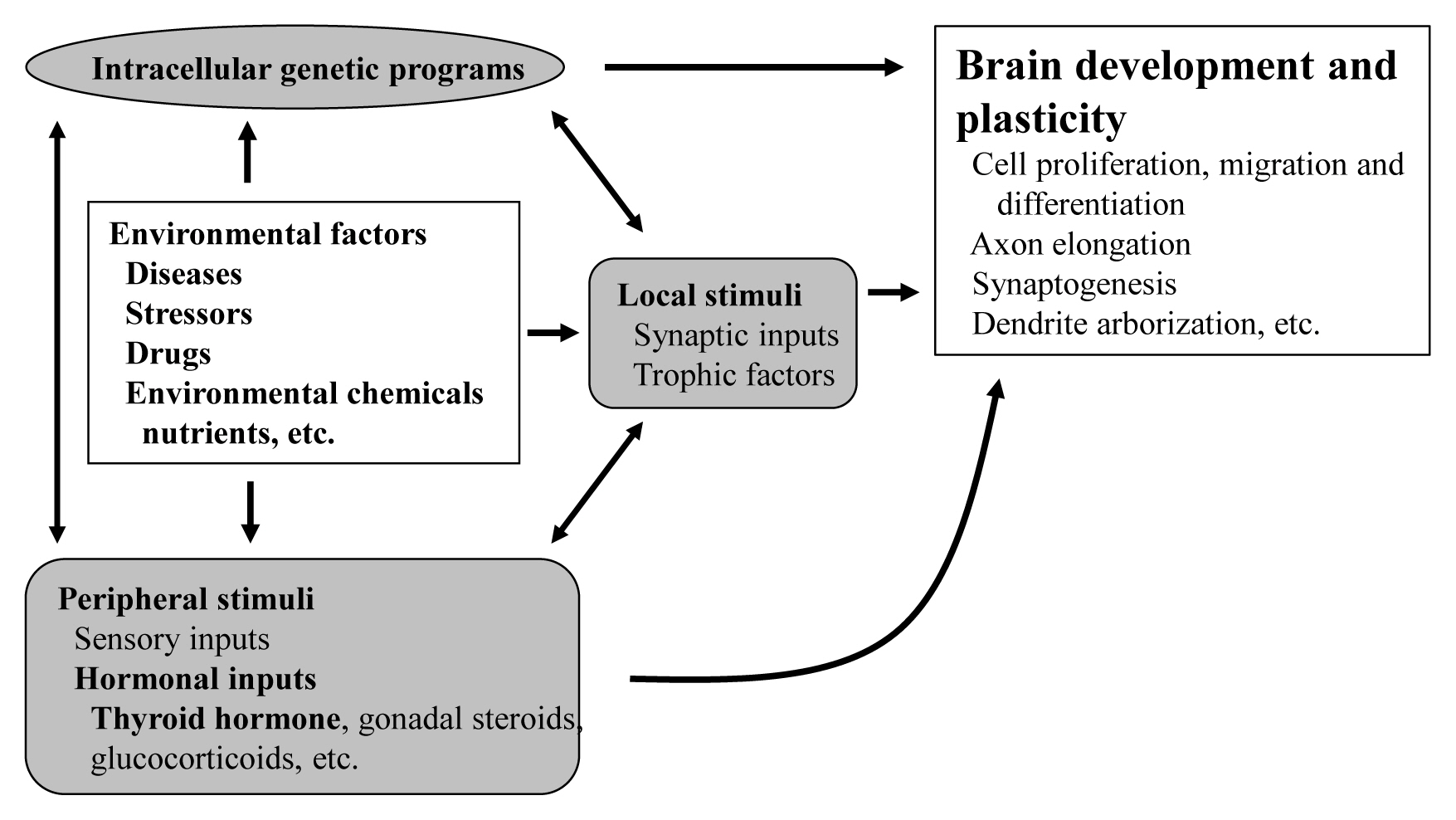
Schematic diagram of multiple factors involved in brain development and plasticity.
The brain consists of complex interneural networks. The cerebellum is one of few sites in the brain in which the neural network has been extensively studied. The structure of its cortex is well-organized, with a highly specific and uniform arrangement of cells and microcircuitry [2]. Moreover, experimental interventions on the cerebellum are easier in rodents given its postnatal organization. Therefore, the cerebellum is an excellent system for the study of neural development and neural plasticity.
As peripheral stimuli, hormones produced in the endocrine cells and released into the blood stream significantly influence the development of the brain including the cerebellum. Some hormone receptors are expressed in the cerebellum. However, the role of hormones in the development and plasticity of the cerebellum is not fully understood.
Among numerous circulating hormones, some small lipophilic hormones including thyroid hormone (TH), gonadal hormones, and glucocorticoids are involved in cerebellar development. These hormones are more capable of crossing the blood-brain barrier (BBB) than peptide hormones because of their chemical properties, although there have been studies incriminating some specific transporters in this process [3]. Receptors for these hormones belong to the nuclear receptor (NR) superfamily and regulate the transcription of the target genes in a ligand-dependent manner [4]. NRs are widely distributed in the CNS with specific expression profiling [5]. Particularly, during cerebellar development, NRs exhibit a temporal and spatial expression pattern [6]. However, the role of NRs during cerebellar development is still under investigation.
Among these lipophilic hormones, THs are well-known major regulators of cerebellar development. In both rodents [7–9] and humans [10], hypothyroidism during the postnatal developmental period results in abnormal morphogenesis or altered function. This review focuses on the role of TH in cerebellar development. Molecular and cellular actions of TH will be described. Furthermore, cerebellar defects in experimental congenital hypothyroidism (CH) are also summarized.
MOLECULAR AND CELLULAR ACTIONS OF TH
THs are synthesized from tyrosine and iodine in the thyroid gland. Thyroid peroxidase and dual oxidase (DUOX) play central roles in the iodine incorporation to tyrosine residues in thyroglobulin [11]. THs include L-triiodothyronine (T3) and L-tetraiodothyronine (thyroxine [T4]). T4 is the main TH produced in the thyroid gland. T3 is produced either directly in the thyroid gland or by deiodination of T4 in the peripheral tissues. Type 2 deiodinase (DIO2) is the dominant enzyme responsible for deiodination in the brain [12]. THs enter the cells largely via membrane transporters such as monocarboxylate transporter 8 (MCT8) [13].
THs bind to the thyroid hormone receptor (TR) in the nuclei and regulate transcription of the target genes [14]. T3 possesses higher affinity to TR and is regarded as active form. As a member of the NR superfamily, TR contains a DNA-binding domain (DBD) and a ligand-binding domain (LBD). The DBD is highly conserved among multiple NRs, whereas the LBD contains the ligand-dependent activation domain, which is also responsible for dimerization of the NRs. The homology of LBD is relatively high among NRs. The highly variable N-terminal region harbors autonomous activation function.
TRs are encoded by two genes, THRA and THRB, which are located on chromosome 17 and 3 in humans and 11 and 14 in mice [15]. These genes generate several TR isoforms (Fig. 2). Three major isoforms, TRα1, TRβ1, and TRβ2, bind TH and serve as ligand-dependent transcriptional factors. These isoforms are functionally similar; however, their roles are distinguished depending on their distinct expression profiles. TRα2 lacks the capacity to bind TH and works as an endogenous inhibitor of other TRs [16]. In addition, some truncated proteins such as TRΔα1 and TRΔα2 are produced. At least in mice, these two proteins have been shown to be functional transcriptional suppressors in vivo [17]. Furthermore, TRβ3 and its related protein were also reported, although these are regarded as minor isoforms [18].

Thyroid hormone receptors and their related proteins generated from THRA or THRB gene. Numerals indicate the number of amino acids. TR, thyroid hormone receptor.
TR exerts bi-directional functions similarly to retinoic acid receptor and vitamin D receptor (Fig. 3) [19]. TR binds to specific nucleotide sequences known as TH response element (TRE) on its target genes as a homodimer, or as a heterodimer with retinoid X receptor. Corepressor complexes bind TR and suppress transcription in the absence of a ligand. An addition of TH induces an exchange of cofactors. Corepressor complexes are released from TR and coactivator complexes are recruited, which stimulates transcription. Cofactors including corepressors or coactivators may alter chromatin structure by modulating histone modification or the stability of the basal transcriptional machinery [14,20]. Recently, it was reported that the cofactor exchange does not follow such a canonical “all-or none” switch model in vivo; rather, the expression of TR-target genes is actually regulated by a shift in the relative binding of corepressors and coactivators [21].
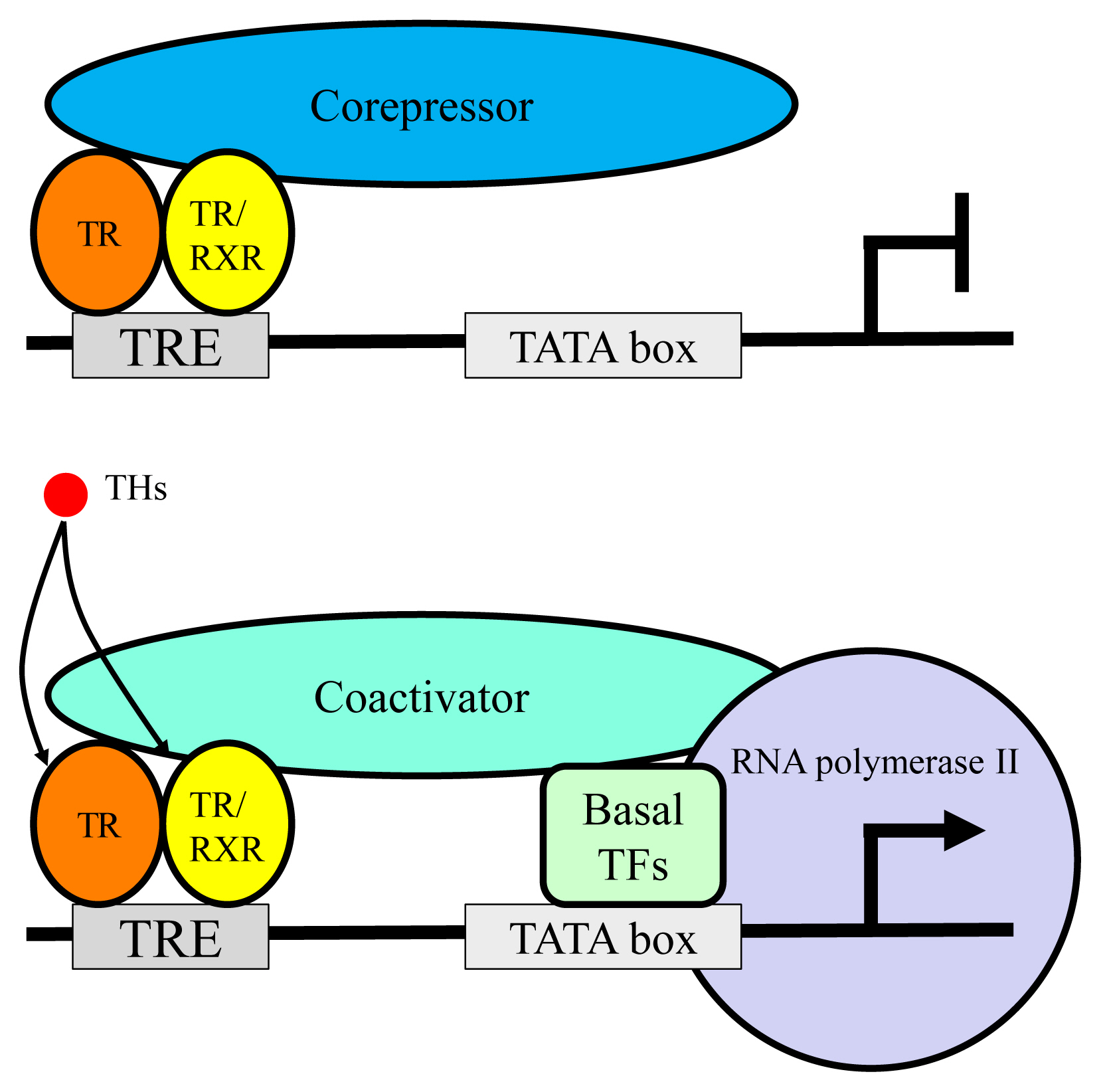
Schematic figure showing the mechanisms of thyroid hormone receptor (TR)-mediated transcription. RXR, retinoid X receptor; TRE, thyroid hormone response element; TF, transcriptional factor.
The upregulated target genes are responsible for the cellular effects of THs. These effects include some basic cellular functions such as cell proliferation, migration, and differentiation, which are necessary for brain development and plasticity (Fig. 1). It is suggested that non-genomic action of TH through membranous integrin αvβ3 [22] is responsible for proliferation of cells [23], although TR-target genes responsible for cell proliferation are not yet fully established. TH induces epithelial-mesenchymal transition and stimulates migration by upregulating the zinc finger E-box binding homeobox 1 (ZEB1) gene in squamous cell carcinoma cells [24]. Furthermore, TH plays important roles in the maturation and differentiation of numerous types of cells including neurons and glial cells. Neural differentiation by TH contributes to multiple phenomena such as axon elongation, synaptogenesis, and dendrite arborization. Some examples of TR-target genes responsible for cerebellar development are Purkinje cell protein-2 and myelin basic protein [25]. In addition, the upregulated TR-target genes further induce secondary or tertiary effects. The whole picture of TH-induced cerebellar development remains to be clarified.
ANIMAL MODEL FOR RESEARCH ON TH ACTION IN THE CEREBELLUM
TH deficiency during the postnatal period causes CH in humans. Untreated CH causes poor neurodevelopmental outcomes in the children such as mental retardation, cerebellar ataxia, and deafness, together with impaired body growth [26,27]. Thus, the cerebellum has been identified as a TH-sensitive brain region during the developmental age. Various kinds of mice models have been used to assess the effects of TH on the development of the cerebellum (Table 1) [28–52]. These are categorized to drug-induced hypothyroidism, surgical thyroidectomy, or genetic mutation. These models display extensive abnormalities in cerebellar development, resulting in an ataxic phenotype based on morphological or neurophysiological impairment.
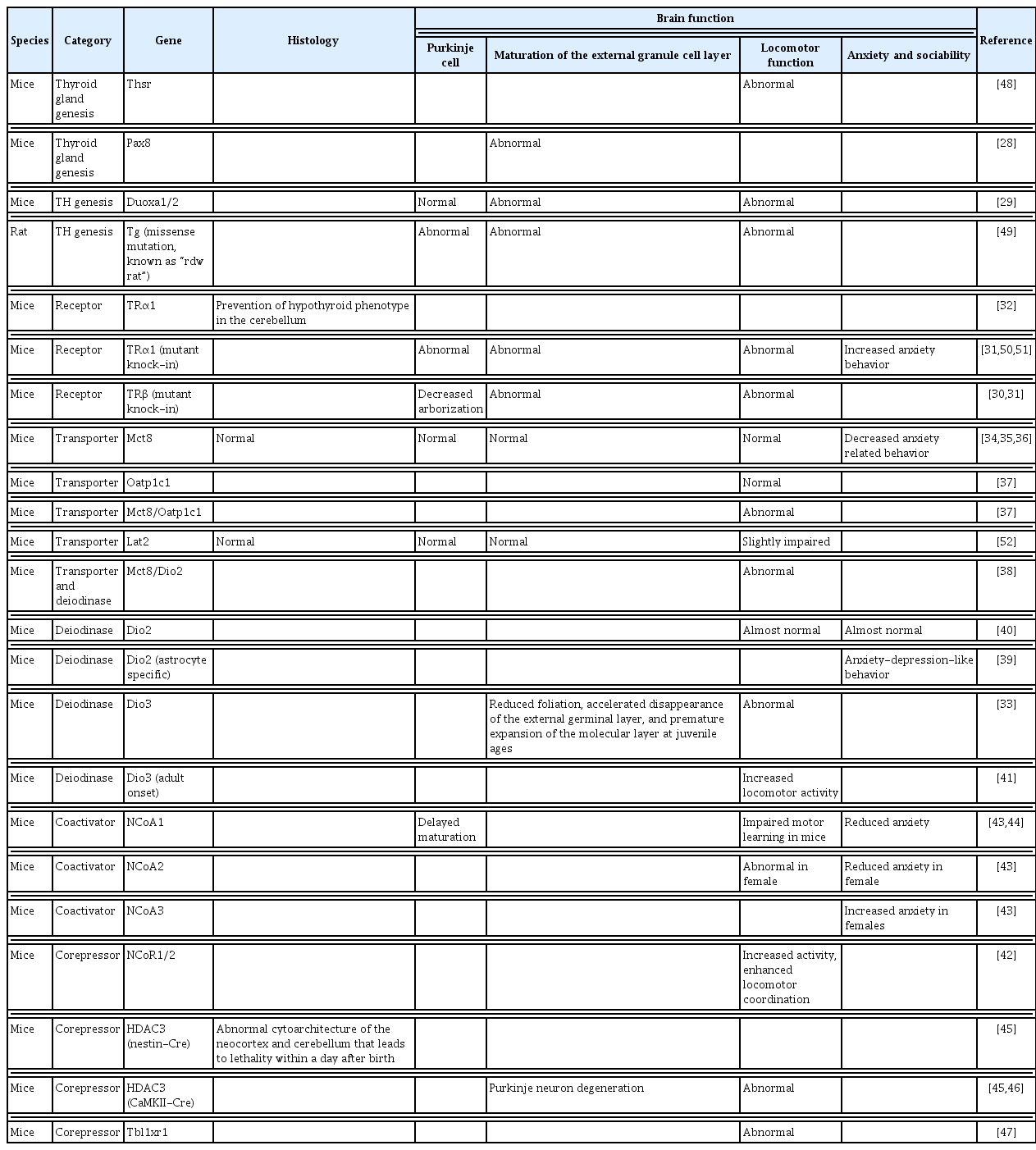
Representative Phenotype of Animal Models for the Study of the Action of Thyroid Hormone in the Cerebellum
DEFECT IN THYROID DYSGENESIS AND THYROID DYSHORMONOGENESIS
One of the major causes of primary hypothyroidism is the abnormalities in thyroid gland development (thyroid dysgenesis). The other cause is errors of TH genesis (dyshormonogenesis). The most common CH rodent models are the drug-induced models with anti-thyroid drugs such as propylthiouracil or 2-mercapto-1-methylimidazole (MMI) [53,54]. Nicholson and Altman reported a reduced cerebellar weight; a prolonged cell proliferation in the external granule cell layer (EGL) and retarded EGL disappearance; a retarded cell differentiation in the molecular and internal granule cell layers, and terminal increases in the numbers of granule cells and astrocytes; and a decrease in the numbers of basket cells. They equally reported a myelination delay, and synaptic disconnections among cerebellar neurons and afferent neuronal fibers from other brain regions [55,56]. Legrand [53] and Clos et al. [57] demonstrated a decreased dendrite arborization of Purkinje cells, two-fold-longer primary dendrites, reduction of growth and branching of dendrites, and shorter parallel fibers with fewer synaptic contacts with Purkinje cells. Such impairment of cerebellar Purkinje cell growth was confirmed by ex vivo or in vitro human or rodent models. Moreover, the drug-induced model can enable the study of the effects of mild hypothyroidism on brain development. Mice with mild perinatal hypothyroid had an impaired cerebellar development, and its effect will span across generations.
In addition to drug-induced hypothyroid animal models, mutations or gene-modifications of several genes that regulate the development of the thyroid gland have been reported as models showing CH. For instance, the paired box 8 (PAX8) gene is required for follicular cells of the thyroid gland for both human and mice [58,59]. Pax8−/− mice showed an organization defect and a reduction of dendrites elaboration that can be rescued by TH treatment [28]. Knockout mice of Duoxa gene (Duoxa−/−), which plays the rate-limiting role of TH synthesis, showed similar morphological changes in the cerebellum at postnatal 2 weeks with severe hypothyroidism. However, cerebellar dysfunction persisted throughout the life although morphological changes had occurred only around the weaning age. Compared to other CH model mice, Duoxa−/− mice may be caused by the combined effect of CH and the functional disruption of the DUOX/DUOXA complex in the brain [29].
DEFECTS IN TH RECEPTOR ACTION AND FUNCTION
Resistance to thyroid hormone (RTH) is the most common category of impaired sensitivity to TH in humans. RTH is a syndrome characterized by reduced intracellular action of T3 and was first identified as a syndrome of reduced end-organ responsiveness to TH in 1967 [60]. About 85% of families with RTHβ harbor mutations in TRβ [61]. Due to the defects in the feedback regulation of the hypothalamic-pituitary-thyroid axis, patients with RTHβ show high levels of T4 and T3 and normal or high levels of thyroid stimulating hormone (TSH). Patients with RTHβ present with various symptoms. Some patients show hyperthyroidism, while others exhibit hypothyroidism. The diversity of the symptoms can be explained by the various responsiveness to the elevated levels of TH. It was reported that patients RTHβ present a high prevalence of attention deficit disorder and learning disabilities, which are similar to the symptoms of hyperthyroidism [62,63]. However, some patients who harbor mutant TRβ with severe dysfunction have the symptoms of CH. Whereas, RTHα was first discovered in 2012 [64]. Since TRα is not involved in the feedback regulation of the hypothalamic-pituitary-thyroid axis, patients with RTHα tend to have low serum T4, borderline high T3, and lower rT3. Major clinical findings regarding the CNS in patients with RTHα are mental retardation and motor impairment, which are similar to the symptoms of CH [65]. However, much is yet to be known in order to characterize the RTHα mutation in humans.
RTH model mice, knock-in mice harboring mutant TRs, have been generated. Mice harboring the Δ337T mutant TRβ phenocopy the hypothyroid symptoms of patients with RTHβ carrying the same mutation [30]. In a similar manner to patients with RTHα, mutant TRα knock-in mice also exhibit a hypothyroid phenotype [31]. The neurological phenotypes of the cerebellum of animal models were abnormal cerebellar development, decreased arborization of Purkinje cell dendrites, and impaired locomotor activity. The cerebellar phenotype of RTH animal models is more severe than that of TR knockout animals described below, indicating that the abnormal cerebellar development seen in hypothyroid animals may be induced mainly by unliganded TRs. Furthermore, the effect may not be a result of generalized TH resistance, but may be because of the cerebellar-cell specific action of TH resistance. This hypothesis is supported by studies using animal models expressing dominant-negative TRs in cerebellar cells, showing aberrant cerebellar development [66,67].
Various TR gene knockout models were also generated; however, these animal models may not always be suitable for studying the action of TH in the cerebellum because of the bi-directional actions of transcriptional regulation of target genes of TR (Fig. 3). Since TR deletion abolishes the repressive action of TR, phenotypes of TR knockout mice are greatly different from those of mice harboring low TH levels. However, TR knockout mice are essential for the study of the role of TR in organ development and function. Another issue that may be considered to generate TR knockout mice is that some introns, such as intron 7 of Thra, have a weak promoter activity. Thus, deleting upstream exons may result in the expression of additional TR-related proteins, which may be limited under normal conditions [68]. As discussed above (Fig. 2), at least three additional TR-related proteins, TRΔα1, TRΔα2, and TRΔβ3, may be generated. Thus, phenotypes of TR knockout mice may result from the combined deletion of a specific TR with overexpression of other TR species. There have also been reports of TRα1-deleted mice showing a limited alterations in behavior and neural circuit [69]. However, except for aberrant maturation of astrocytes, their cerebellar phenotype appeared normal [70]. More strikingly, TRα1 deletion prevented the structural alteration of the cerebellum in hypothyroidism induced by MMI and perchlorate treatment [32]. These results indicate that the abnormal cerebellar phenotype in animals with thyroid dyshormonogenesis may result from the dominant-negative action of unliganded TRα proteins. Interestingly, deleting TRα1 also prevented the structural alteration induced by deleting DIO3 [33]. These results indicate that, liganded TRα1 plays an important role in cerebellar development although the cerebellar phenotype of TRα1 deleted mice is limited. Conversely, TRα2 knockout mice show both hyper- and hypothyroid phenotypes in an organ-specific manner [71]. This may be a result of elevated TRα1 expressions in this mouse. TRα1 expression in the brain is also elevated, but the cerebellar phenotype was unclear. The deletion of both TRα1 and TRα2 also shows only a limited phenotype in the cerebellum. However, besides the cerebellar phenotype, the existence of TRΔα1 and/or TRΔα2 shows altered phenotypes in various organs. When TRα1 and TRα2 are deleted, but expressions of TRΔα1 and TRΔα2 are not inhibited (Thra−/−) [72], their phenotype is more severe than those of mice in which all TRα proteins are deleted (Thra0/0) [17,73]. The decrease in plasma TH levels is greater, and there is a more severe impairment of bone and intestine development. A more limited brain phenotype is observed in TRβ knockout mice, while TRβ1 is widely expressed including in the cerebellum, particularly in Purkinje cells. However, the abnormal brain phenotype seems to be confined to the hypothalamus, and changes in cerebellar phenotype have not been reported [74,75]. Because the function of one receptor cannot be substituted for the other in the case of TRα and β double knockout, their phenotypes are more severe than those of single gene knockout [76,77]. However, there has been no detailed study of the altered brain development in these double knockout mice.
DEFECTS IN TH TRANSPORT
TH transporters are required to pass the BBB and reach to neurons in the CNS. Two major transmembrane transporters, MCT8 and organic anion transporting polypeptide 1C1 (OATP1C1), are known to transport T3 and T4 specifically. OATP1C1 has a high affinity for T4 and rT3, but a low affinity for T3 [78,79]. MCT8 and OATP1C1 are located in neural cells, the endothelial cells of microvessels, and the choroid plexus [80–82]. In particular, MCT8 is essential for transporting TH through the BBB [34,83]. Solute carrier family 16 member 2 (SLC16A2) mutations, encoding MCT8, cause X-linked Allan-Herndon-Dudley syndrome (AHDS). AHDS is characterized by an altered thyroid function (low T4, high T3, and borderline-increased TSH levels) and TH metabolism and a severely impaired neurodevelopment. Solute carrier organic anion transporter family member 1C1 (SLCO1C1) mutations, encoding OATP1C1, have recently been described to express brain-specific hypothyroidism, severe brain hypometabolism, and juvenile neurodegeneration [84].
Mice models have been described to assess the function of TH transporters in the CNS. Initially, MCT8 deficient mice were proposed as an AHDS model. Although Mct8−/− mice showed a thyroid profile similar to that of the patients and relatively low levels of T3 in different brain areas, there were no major defects in brain histopathology, nor few behavioral changes [35,36,85]. In particular, Mct8−/− mice revealed no effects on Purkinje cells morphology in the cerebellum [35]. The differences between humans and mice suggest that Mct8−/− mice may have a compensatory mechanism to supply TH to neurons with different factors such as OATP1C1 and DIO2. Interestingly, double knock out of Mct8/Oatp1c1 revealed low uptake of both T3 and T4 into the brain, delayed cerebellar development, and reduced myelination with reduced locomotor activities [37]. Mct8/Dio2 double knockout mice also presented as a model for human MCT8 deficiency, with brain hypothyroidism, alterations in histology, and impaired motor skills [38]. Thus, Mct8 deficiency in mice is compensated by T4 transport through the OATP1C1. This hypothesis is supported by the higher expression of OATP1C1 in rodent BBB compared to that in humans [80,81]. The necessity of MCT8 is also confirmed in the developing chicken cerebellum. The inactivation of MCT8 revealed smaller and less complexed dendritic trees of Purkinje cells, disruption of granule cell precursor proliferation, and post-mitotic granule cell maturation [86]. Recently, the expression pattern of MCT8 appeared to be spatiotemporally changed in the brains of humans and mice [87]. During postnatal development, MCT8 was expressed stably in the endothelial cells of the BBB, choroid plexus epithelial cells, and tanycytes. Conversely, it was robustly detectable in specific brain regions including the cerebellum of young mice and strongly declined with age [87]. These results suggest that spatiotemporal expression patterns of MCT8 contribute to the high TH demands in the developing brain.
DEFECTS IN LOCAL TH METABOLISM
Iodothyronine deiodinase enzymes play important roles for activation and deactivation of TH. In the CNS, DIO2 is predominantly expressed in astrocyte, and converts prohormone T4 to the active hormone T3 [39]. In contrast, DIO3 is mainly expressed in the neuron, and converts T4 to rT3 and T3 to T2 for inactivating THs [88]. It is suggested that the balance between the enzyme activity of DIO2 and DIO3 may determine the local concentration of T3. The balance of DIO2 and DIO3 are known to be controlled dynamically in the rat developing cerebellum [89]. In the second postnatal week, DIO2 activity is upregulated, whereas DIO3 activity is downregulated [90]. These changes correlate with the THs demands, such as myelination and synaptogenesis, in a sensitive period in the mouse cerebellum. Several knockout mice were generated to determine the role of DIO2 and DIO3 in the brain. The Dio2−/− mice was expected to show a similar phenotype to severe global hypothyroid mice. However, neurological functions in Dio2−/− mice were only slightly changed, although the level of T3 was decreased in the cerebellum [40]. Whereas, systemic and astrocyte-specific Dio2−/− mice exhibited increased anxiety and fear memory [39,91]. These reports suggest that compensatory mechanisms protect T3-dependent responses in Dio2−/− mice. This hypothesis was supported by the result of a double knockout of Dio2/Mct8. Mice lacking both Dio2 and Mct8 demonstrated motor skill impairments [38]. In contrast, Dio3−/− mice displayed increased T3-dependent gene expression in the brain suggesting increased of T3 concentration [41,92,93]. Dio3−/− mice exhibited reduced foliation, accelerated disappearance of the EGL, and premature expansion of the molecular layer at juvenile ages [33]. Dio3 knockout mice also exhibited locomotor behavioral abnormalities and impaired ability in descending a vertical pole. Furthermore, double deletion of Dio3 and Tra1 substantially corrected the cerebellar and behavioral phenotypes [33]. Dio3−/− mice also exhibit hyperactivity, decreased anxiety-like behavior, and lack of maternal behavior in female mice [33,94]. These mice models help in understanding that an appropriate balance of DIO2/3 is required in developing brain.
DEFECTS IN COREGULATORS IN TH ACTION
The action of TH is mediated by TR and several transcriptional factors. NR coactivators (NCoAs) and corepressors (NCoRs) bind to NRs including TR in a ligand-dependent manner and mediate the transcriptional activities. Nuclear cofactors are known to be expressed in the CNS; however, little is known about their neuronal physiological roles. Recently, reports of genetic variants of nuclear cofactors in CNS-related conditions in humans have begun to emerge [95]. The p160 steroid receptor coactivator (SRC) family comprises three homologous genes: SRC-1 (NCoA1), SRC-2 (NCoA2), and SRC-3 (NCoA3). Heterozygous missense NCoA1 variants found in severely obese individuals impair leptin-mediated proopiomelanocortin (Pomc) reporter activity in humans. Moreover, de novo genetic variants in NCoR1, NCoR2, and histone deacetylase 3 (HDAC3) are found in pediatric patients with intellectual disabilities or autism spectrum disorders (ASD) [42,96–99]. Leptin-induced depolarization of Pomc neurons and Pomc expression were reduced, and food intake and body weight were increased in a knock-in mouse model of a loss of function human variant (SRC-1L1376P) [100]. In the developing rat cerebellum, NCoA1 is predominantly expressed in Purkinje cells and granule cells in the internal granule cell layer. The level of NCoA1 protein in the cerebellum was greatest at P15, when the action of TH may be obvious. The differential expression of SRC-1 may be crucial in mediating the action of TH during cerebellar development [101].
Mutation each NCoA family showed different phenotypes in mice. Male NCoA1−/− mice had motor learning impairment, while females NCoA2−/− mice had impaired motor coordination. However, NCoA3−/− mice showed no changes on motor skills [43,44]. NCoA2−/− females also showed decreased anxiety, whereas NCoA3−/− female mice had increased anxiety and reduced exploratory activity and impairments in prepulse inhibition [43]. NS-DADm mice, specifically NCoR1/2, abolished their ability to activate HDAC3. These mice showed increased activity, enhanced locomotor coordination, reduced anxiety, impaired social interaction, and impaired spatial learning and recognition memory [42]. Conditional deletion of HDAC3 in mice in generated using nestin-Cre caused abnormal cytoarchitecture of the neocortex and cerebellum, leading to lethality within a day after birth [45]. Another HDAC3 conditional knockout mice using Camk2a-Cre demonstrated a normal organization of cerebellar Purkinje neurons at birth. However, these mice displayed progressive hindlimb paralysis, ataxia, higher numbers of astrocytes, and Purkinje neuron degeneration, which leads to lethality at approximately 6 weeks of age [45,46]. Collectively, both NCoA and NCoR have unique brain regions and gender specific functions.
Transducin beta-like 1 X-linked (TBL1X) and TBL1X receptor 1 (TBL1XR1), which are the members of HDAC-containing NCoRs complexes, play essential roles in the development of the brain. Targeted sequencing of over 200 genes in more than 10,000 patients with ASD, intellectual disability, seizure, microcephaly, or macrocephaly identified 13 cases carrying disruptive mutations in TBL1XR1 [102]. Moreover, patients with TBL1XR1 mutations display compromised or delayed motor skills [103]. Tbl1xr1−/− mice revealed impaired motor coordination, memory skills, and social activity. Such phenotypes were also described at different levels in patients harboring this mutation [47,103, 104]. TBL1X defects are associated with central hypothyroidism and hearing loss [105,106]. Genome-wide association studies has revealed that TBLX1 is also linked to autism. However, neurodevelopmental defects have not yet been reported in these patients [107]. Further studies are required to identify the role of TBL1XR1 and TBL1X in the developing brain.
In addition to NcoAs and NcoRs, TR may interact with other NRs that regulate gene expression such as retinoic acid related orphan receptor α (RORα). RORα is strongly expressed in Purkinje cells, and plays a critical role in cerebellar development. Cerebellar phenotype and alteration of neurotrophin expression of natural mutant mouse (staggerer) harboring an RORα mutation is similar to that in the hypothyroid mouse [108], although its thyroid function is normal. This indicates that RORα may be involved in TH-regulated gene expression in the developing cerebellum. In fact, TH regulates RORα expression during the first two postnatal weeks [54,66], indicating that TH may alter gene expression critical for cerebellar development through RORα regulation. Furthermore, RORα augments TR-mediated transcription, whereas staggerer-type mutant RORα does not have such action [108]. Another study showed that RORα may directly interact with TR without binding to TRE. The DBD of RORα may play a role in such an interaction [109]. These results indicate that RORα is required for full TR function in the developing cerebellum. Although there is still little evidence of the mechanism by which transcriptional factors contribute to TH signaling in brain, each transcriptional factor plays an important role in the development of the brain.
CONCLUSIONS
It is established that THs play a pivotal role in cerebellar development. In addition, the molecular mechanisms of the action of TH have been extensively studied. However, the mechanisms through which THs regulate cerebellar development are still under investigation. Further studies including behavioral, neurophysiological, morphological, and molecular aspects are necessary to reveal the whole picture of TH-dependent cerebellar development.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was funded by JSPS KAKENHI Grant No. 21K08569 to Sumiyasu Ishii, Grant-in-Aid for Early-Career Scientists (Grant 19K16486 and 21K15340 to Izuki Amano), and Grant-in-Aid for Scientific Research (B) (Grant 18H03379 to Noriyuki Koibuchi) from the Japan Society for the Promotion of Sciences.