
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 36(1); 2021 > Article
-
Review ArticleThe Genotype-Based Morphology of Aldosterone-Producing Adrenocortical Disorders and Their Association with Aging
-
Xin Gao1
 , Yuto Yamazaki1, Yuta Tezuka2,3, Kei Omata2,3, Yoshikiyo Ono3, Ryo Morimoto3, Yasuhiro Nakamura4, Fumitoshi Satoh2,3, Hironobu Sasano1
, Yuto Yamazaki1, Yuta Tezuka2,3, Kei Omata2,3, Yoshikiyo Ono3, Ryo Morimoto3, Yasuhiro Nakamura4, Fumitoshi Satoh2,3, Hironobu Sasano1 -
Endocrinology and Metabolism 2021;36(1):12-21.
DOI: https://doi.org/10.3803/EnM.2021.101
Published online: February 24, 2021
1Department of Pathology, Tohoku University Graduate School of Medicine, Sendai, Japan
2Division of Clinical Hypertension, Endocrinology and Metabolism, Tohoku University Graduate School of Medicine, Sendai, Japan
3Division of Nephrology, Endocrinology, and Vascular Medicine, Tohoku University Hospital, Sendai, Japan
4Division of Pathology, Faculty of Medicine, Tohoku Medical and Pharmaceutical University, Sendai, Japan
- Corresponding author: Hironobu Sasano. Department of Pathology, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Aoba-ku, Sendai 980-8575, Japan, Tel: +81-22-717-8050, Fax: +81-22-717-8051, E-mail: hsasano@patholo2.med.tohoku.ac.jp
Copyright © 2021 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- GENETIC ALTERATIONS ASSOCIATED WITH PRIMARY ALDOSTERONISM
- THE GENETIC SPECTRUM OF THE HISTOLOGICAL SUBTYPES OF PA
- HISTOPATHOLOGY AND CELLULAR MORPHOLOGY OF PA
- POTENTIAL ASSOCIATION OF APM AND APA
- SUBCLINICAL OR NORMOTENSIVE PA
- AGING AND ALDOSTERONE
- CONCLUSIONS
- Article information
- References
ABSTRACT
- Primary aldosteronism (PA) is the most common cause of secondary hypertension, and is associated with an increased incidence of cardiovascular events. PA itself is clinically classified into the following two types: unilateral PA, mostly composed of aldosterone-producing adenoma (APA); and bilateral hyperaldosteronism, consisting of multiple aldosterone-producing micronodules (APMs) and aldosterone-producing diffuse hyperplasia. Histopathologically, those disorders above are all composed of compact and clear cells. The cellular morphology in the above-mentioned aldosterone-producing disorders has been recently reported to be closely correlated with patterns of somatic mutations of ion channels including KCNJ5, CACNA1D, ATP1A1, ATP2B3, and others. In addition, in non-pathological adrenal glands, APMs are frequently detected regardless of the status of the renin-angiotensin-aldosterone system (RAAS). Aldosterone-producing nodules have been also proposed as non-neoplastic nodules that can be identified by hematoxylin and eosin staining. These non-neoplastic CYP11B2-positive nodules could represent possible precursors of APAs possibly due to the presence of somatic mutations. On the other hand, aging itself also plays a pivotal role in the development of aldosterone-producing lesions. For instance, the number of APMs was also reported to increase with aging. Therefore, recent studies indicated the novel classification of PA into normotensive PA (RAAS-independent APM) and clinically overt PA.
- Primary aldosteronism (PA) accounts for 5% to 10% of hypertensive patients, usually accompanied with increased cardiovascular events including stroke, ventricular hypertrophy, fibrosis, vascular remodeling, and others [1–4]. PA is mainly classified into neoplastic and non-neoplastic lesions based on the biological characteristics of lesions responsible for aldosterone excess. However, the histopathological identification of these lesions by hematoxylin and eosin (H&E) staining (morphology) is well known to be extremely difficult even when differentiating between neoplasms and non-neoplastic nodules. Due to the development of cytochrome P450 family 11 subfamily B member 2 (CYP11B2) immunohistochemistry, rather unexpected patterns of CYP11B2 have been revealed [5]. In contrast to the diffuse CYP11B2-positive cells in ZG, CYP11B2-positive and cytochrome P450 family 11 subfamily B member 1 (CYP11B1)- and cytochrome P450 family 17 subfamily A member 1 (CYP17A1)-negative adrenocortical cells were identified as clusters of CYP11B2-positive cells in the subcapsular areas previously termed as aldosterone-producing cluster cells (APCCs). Various nomenclature systems for the histopathological classification of aldosterone-producing lesions have been proposed at this juncture, but the international consensus of PA histopathology was very recently published, and the terms were re-defined and integrated [6].
- PA is clinically classified into two subtypes, unilateral hyperaldosteronism (UHA) and bilateral hyperaldosteronism (BHA), according to the results of adrenal vein sampling [7,8]. In addition, UHA is histologically classified into aldosterone-producing adenoma (APA) and aldosterone-producing micronodules (APMs) using CYP11B2 immunohistochemistry [6,9]. BHA includes multiple APMs and aldosterone-producing diffuse hyperplasia (APDH) and occasionally bilateral APAs [6,10]. Those non-neoplastic nodules involved in aldosterone overproduction were also recently reported to harbor APA-driver somatic mutations of various ion channels including potassium inwardly rectifying channel subfamily J member 5 (KCNJ5), calcium voltage-gated channel subunit alpha1 D (CACNA1D), ATPase Na+/K+ transporting subunit alpha 1 (ATP1A1), and ATPase plasma membrane Ca2+ transporting 3 (ATP2B3), which indicated that APM could represent the precursor lesion of APA [11,12]. In addition, the number of APMs was significantly positively correlated with aging of the subjects, although serum aldosterone level usually declines with aging [11,13]. Therefore, we herein review recent developments regarding the pathogenesis of PA and its association with aging and histopathological features of the lesions.
INTRODUCTION
- PA is caused by autonomous aldosterone production due to either hyperplasia or nodules. This aldosterone overproduction was demonstrated to be caused by genetic mutations encoding for various ion channels such as KCNJ5, CACNA1D, ATP1A1, and ATP2B3 [14–17]. All of those genes above are well known to regulate intracellular ionic homeostasis and cell membrane potential, resulting in increased intracellular calcium levels and subsequently promoting aldosterone biosynthesis [14–17]. In addition, rare PA cases were reported to harbor somatic mutations of catenin beta 1 (CTNNB1) and chloride voltage-gated channel 2 (CLCN2) encoding for β-catenin and the chloride channel ClC-2 [18,19].
- KCNJ5 mutation is the most frequent somatic mutation reported in APA, although its prevalence differs among ethnicities [14,20]. KCNJ5 encodes inward rectifier K+ channel 4, and its mutations cause increased Na+ permeability, subsequently resulting in a sustained depolarization of the cell membrane [14]. Aldosterone biosynthesis could also be promoted by increased cytosolic calcium levels through activated calcium channels by increased intracellular K+ concentrations [21].
- CACNA1D encodes L-type calcium channel α-subunit CaV1.3, which regulates intracellular calcium homeostasis [22]. Mutation of CaV1.3 also stimulated an influx of Ca2+, resulting in autonomous aldosterone production [16]. Mutations of ATP1A1, encoding α1 subunit of the Na+/K+-ATPase, facilitated an influx of calcium by altering sodium and potassium homeostasis [15]. Mutation of ATP2B3 directly influenced the calcium pump PMCA3, increasing cytosolic calcium levels [23]. Both mutations could result in aldosterone overproduction by increasing intracellular calcium levels.
- Wnt is secreted protein, which regulates growth and stem cell renewal, and its signaling pathway has been well known to be activated in APAs [18]. The Wnt signaling pathway could activate β-catenin, which subsequently promotes the cell proliferation. In addition, mutations of the CTNNB1 gene coding for β-catenin were recently reported in APAs [18]. CTNNB1 mutations were also reported to lead to constitutive activation of β-catenin protein eventually resulting in tumorigenesis, and this mutation is not only restricted to APAs but has also been identified in cortisol-producing adenomas (CPAs) and adrenocortical carcinomas [24].
- A germline calcium voltage-gated channel subunit alpha1 H (CACNA1H) mutation was reported to encode an abnormal voltage-dependent T-type channel CaV3.2, which could induce early-onset hyperaldosteronism and hypertension, although rare [25]. A mutation of CACNA1H is well known to be the main cause of familial hyperaldosteronism (FH) type IV [26]. FH is frequently caused by germline mutations of ion channels such as KCNJ5, CACNA1D, and CACNA1H and further classified into types I–IV according to its clinical and biochemical features [16,26–28]. FH-I is also characterized by glucocorticoid-remediable aldosteronism caused by crossover of both CYP11B2 and CYP11B1 genes, resulting in ectopic expression of CYP11B2 [29,30]. The etiology of FH-II, however, has remained unknown, although its possible association with germline CLCN2 mutations was previously proposed [31]. CLCN2 mutations induced a loss of the voltagegating of the channel, leading to increased chloride influx, which subsequently resulted in cell membrane depolarization [32]. Intracellular calcium levels were eventually increased by depolarization of the cell membrane, as discussed above. On the other hand, FH-III is characterized by development of massive BHA as a result of germline mutations of KCNJ5 [14]. FH-IV is characterized by germline mutations of CACNA1D and CACNA1H [16,25,26].
GENETIC ALTERATIONS ASSOCIATED WITH PRIMARY ALDOSTERONISM
- Aldosterone-producing adenoma
- In APAs, somatic mutations were detected at 88% to 90% of patients. Among those, KCNJ5 is the most frequent genotype regardless of ethnicity. However, 38% of Caucasian patients were reported to harbor KCNJ5 mutations [22,33,34], but a prevalence of 70% was reported among Japanese patients [35]. CACNA1D mutations are the next frequent genotype following KCNJ5 mutations, and 9.3% of European and 2.5% of Japanese APA patients were reported to harbor this somatic mutation in the tumor tissue [22,35]. Mutations of ATP1A1, ATP2B3, and CTNNB1 were rare, constituting approximately less than 5% of APAs regardless of ethnicity [18,22,35].
- Aldosterone-producing non-neoplastic nodules
- These non-neoplastic nodules were reported to harbor somatic mutations like those of APAs [10,11,36,37]. We previously reported somatic mutations in 21 of 61 nodules (34%) from normally-functioning adrenal glands with normotension [11]. Among 21 mutated nodules, 14 harbored CACNA1D mutations; three had ATP2B3 mutations, two had ATP1A1 mutations, and two had both CACNA1D and ATP2B3 mutations. Eight of 23 nodules (35%) were also reported to harbor somatic mutations (six CACAN1D and two ATP1A1) in normal adrenal glands [37]. In addition, another study reported somatic mutations of KCNJ5 in five APMs in adjacent non-pathological adrenal glands [34].
- Non-neoplastic CYP11B2 positive nodules in BHA also frequently harbor somatic mutations of the genes above. We previously reported that 58% of 99 nodules in clinically diagnosed BHA cases harbored CACNA1D mutations and 1% had a KCNJ5 mutation [9]. We also revealed in another study that 65% had CACNA1D mutations, 8% KCNJ5 mutations, and 4% both ATP1A1 and ATP2B3 mutations among a total of 21 nodules identified in the adrenal glands of BHA patients [10]. Of particular interest, no somatic mutations of any of the genes above were detected in the CYP11B2 positive non-nodular hyperplastic zona glomerulosa cells in APDH [10]. These results indicated that there were substantial differences in the etiology and pathogenesis of autonomous aldosterone production between hyperplastic zona glomerulosa cells exhibiting nodular and non-nodular growth, but further investigations are warranted to clarify the details.
THE GENETIC SPECTRUM OF THE HISTOLOGICAL SUBTYPES OF PA
Non-neoplastic nodules in non-pathological adrenal glands
Non-neoplastic nodules in pathological adrenal glands harboring PA
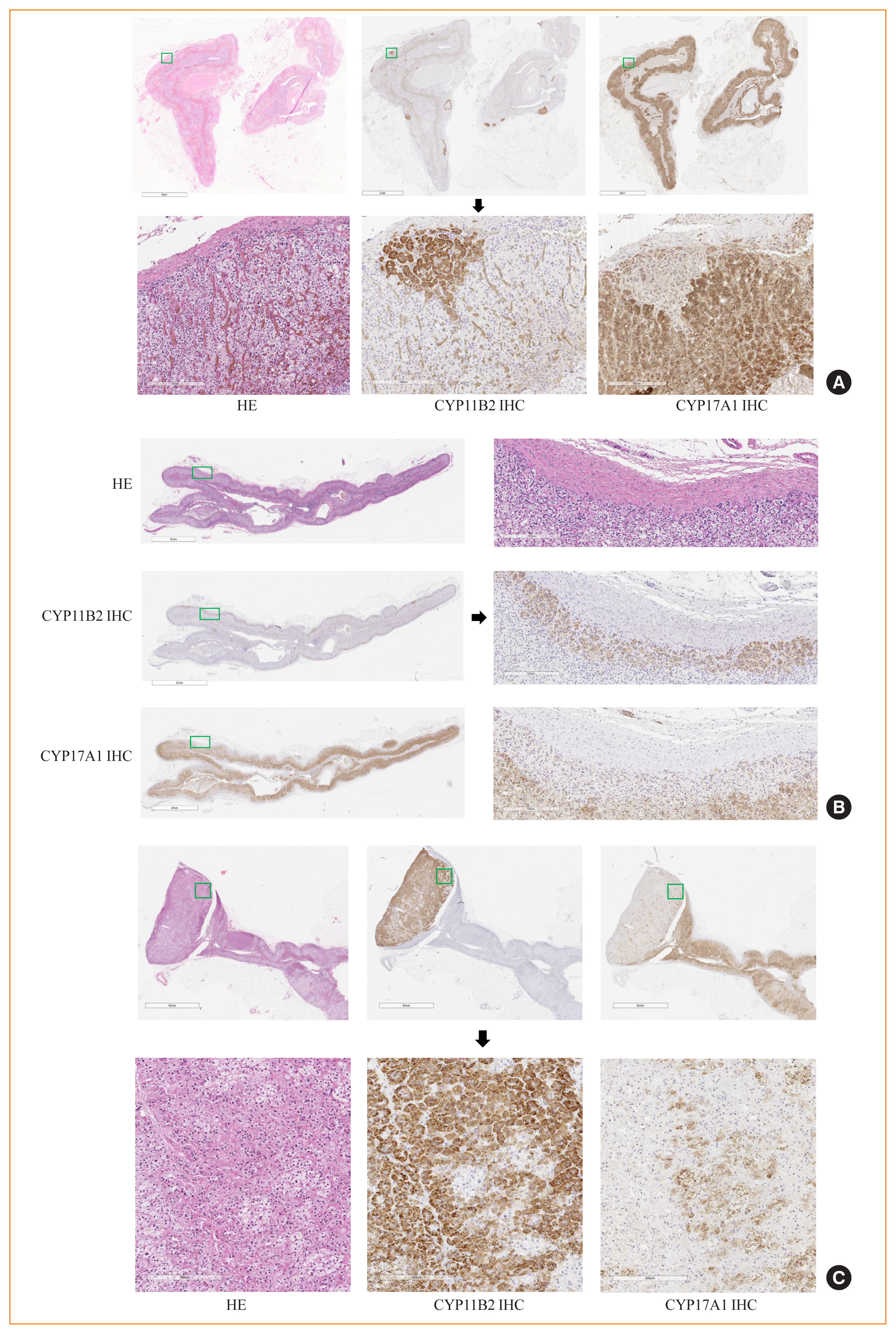
- Adrenocortical cells are basically classified into compact and clear cells, also termed ZG/ZF-like cells from the standpoint of histology [38–41]. Compact cells have a high nuclear to cytoplasm ratio with an eosinophilic lipid-poor cytoplasm containing relatively abundant cell organelles, including mitochondria. On the other hand, clear cells have a low nuclear to cytoplasm ratio with a lipid-rich cytoplasm containing numerous lipid droplets. However, the correlation between these morphological features and functional aspects has remained rather unexplored. Recent developments regarding the relationship between these morphological features and functional findings of PA lesions are summarized in Figs. 1, 2.
- Aldosterone-producing adenomas
- APAs are solitary neoplasms usually measuring more than 10 mm in their greatest dimension and histologically composed of both compact and clear cells with immense intratumoral heterogeneity in their morphological features. The definition of APAs should be based upon both clinical information and histopathological features, including CYP11B2-positive immunoreactivity [6]. We previously reported that KCNJ5 mutated APAs mostly consisted of clear cells [38]. CYP11B2 immunoreactivity was significantly positively correlated with clear phenotype of tumor cells, especially in KCNJ5 mutated APAs [38]. KNCJ5-mutated APAs were also significantly larger than wild-type (WT) ones, consistent with results that KCNJ5-mutated APAs were predominantly composed of more abundant clear tumor cells. We also compared ultrastructural features between KCNJ5-mutated and WT APAs in order to further explore the details of endocrine features between compact and clear cell tumor cells [38]. In WT APAs, a rather low intracellular density of lipid droplets and smaller but more mitochondria were identified by electron microscopic evaluations. In contrast, relatively larger mitochondria with a small number and higher density of lipid droplets were detected in KCNJ5-mutated APAs [38]. This implied that hormonal reactivity in clear cells of KCNJ5-mutated APAs was higher than that of WT ones. We also demonstrated in another study that both ATP1A1- and CACNA1D-mutated APAs harbored a higher ratio of compact tumor cells than clear ones [42]. ATP2B3-mutated APAs were predominantly composed of clear tumor cells, as in KCNJ5-mutated APAs [42]. In addition, CYP17A1 immunoreactivity was more abundant in KCNJ5-mutated APAs, which indicated that KCNJ5-mutated APAs were more active in their hormonal activities synthesizing both aldosterone and cortisol [42]. However, it is also true that intratumoral heterogeneity of compact and clear tumor cells within the same adrenocortical cells harboring the same genotypes has remained unexplored. mRNA expression levels of both CYP17A1 and CYP11B1 were also reported to be significantly higher in compact tumor cells than in clear ones, whereas that of CYP11B2 tended to be higher in clear tumor cells regardless of genotype [43]. This is consistent with results of our previous study that the immunoreactivity of CYP11B1 and CYP17A1 was significantly higher in compact tumor cells of CPAs [39]. Therefore, the hybrid steroidogenesis or simultaneous biosynthesis of both glucocorticoids and mineralocorticoids in the same tumor cells of APAs represented the zonation-deviated phenotype and compact or ZG-like cells had higher cortisol-producing ability, whereas clear or ZF-like cells harbored higher aldosterone-producing ability.
- Based on the statement of international consensus on PA histopathology, aldosterone-producing non-neoplastic nodules are further classified into APMs and aldosterone-producing nodules (APNs) according to whether the nodule is visible (detected) by H&E section (APN was defined as visible nodules) or not, although both types preserve the polarity of zonation, including the gradient of the pattern of CYP11B2 immunoreactivity from the outer to inner part [6]. The previously proposed classifications of “APCC” and/or “micronodule” have been reported to be difficult to distinguish from each other by histopathological findings including CYP11B2 immunohistochemistry alone, and required relevant clinical information such as the presence or absence of hypertension and/or PA. However, the recently proposed international consensus has made it possible to distinguish those non-neoplastic nodules (APM and APN) by histopathological findings alone even without patients’ clinical information. However, we should note that the threshold between APM and APN remains unclear and further discussion is definitively warranted to clearly distinguish these entities from each other. A further improved classification, combined with the clinical information listed above, should clarify the difference between the “physiological” and “pathological” status of those aldosterone-producing adrenocortical non-neoplastic lesions.
- In the normal adrenal gland, CYP11B2 is intensely immunolocalized in the subcapsular area and its immunoreactivity gradually decreases [9,44]. This polarization of aldosterone production detected in normal adrenal glands is maintained in APMs but lost in APAs possible due to the nature of their neoplastic transformation [10,44]. This could contribute to the differential diagnosis of neoplasms versus non-neoplastic nodules in the resected adrenal glands of PA patients. Neither CYP11B1 nor CYP17A1 was expressed, indicating the preservation of development of adrenocortical differentiation. APMs are present not only in the physiologically normal adrenal gland, but of particular interest also in adjacent non-neoplastic adrenal glands with suppression of the renin-angiotensin-aldosterone system (RAAS) of PA patients [5,45]. In addition, APMs in normal adrenal glands also harbored APA-driver somatic mutations as described above, implying their association with APAs.
- APMs or APNs could be detected in BHA cases with multiple numbers and in unilateral PA. In our previous study, 32 nodules were examined in clinically identified BHA cases [10]. Eighteen nodules were composed predominantly of clear cells, 13 of compact cells, and one did not have the information available [10]. The ratio of clear and compact cells was significantly lower in APMs than that in APAs. This is possibly due to the genetic effects on cellular morphology, as KCNJ5 mutations are closely associated with the development of clear cell morphology in autonomous aldosterone-producing adrenocortical lesions and CACNA1D mutations are closely associated with that of compact cells as described above. In clinical practice, the best practical approach to differentiate between APA and non-neoplastic nodule is based on their diameters because of the reasons above, but CYP11B2 immunohistochemistry could also contribute to this differentiation in routine clinical settings.
HISTOPATHOLOGY AND CELLULAR MORPHOLOGY OF PA
Aldosterone-producing non-neoplastic nodules (APMs and APNs)
- APMs have been reported to harbor APA-driver mutations and APMs have been proposed as potential precursors in the development of APA, in addition to the fact that non-neoplastic CYP11B2-positive nodules in adrenal glands in PA patients could be developed from physiological ones, acquiring the ability of excessive autonomous aldosterone production in patients with BHAs due to the predominant genotype of CACNA1D in both entities and the presence of similar histological features [10,12].
- In general, some tumors are considered to originate from stem cells with increased ability of self-renewal following oncogene activation. These stem cells in adrenal glands are considered responsible for continuous replenishment of steroidogenic cells in order to compensate for the cell death required to maintain normal adrenal size and function [46]. Adrenal stem/precursor cells have been detected in both APAs and adjacent ZG cells with weak sonic hedgehog signaling molecule (SHH) expression, which is one of the precursor markers and activated β-catenin indicating their origin from adrenocortical stem cells [46]. ZG cells not only produce aldosterone but also have a relatively high capacity of cellular renewal, and this renewal process follows a centripetal migration: from ZG to ZF [47,48]. In addition, CYP11B2-positive ZG cells can be also differentiated into ZF and CYP11B2 and CYP11B1 double-positive cells present at the ZG-ZF boundary, although extremely rare, resembling the hybrid steroidogenesis in APA cells [48]. In addition, melanocortin 2 receptor (MC2R) expression was detected in APMs, indicating that these cells could potentially differentiate into ZF cells through the protein kinase A signaling pathway [48]. MC2R was also reported to regulate SHH signaling, which further demonstrated its potential differentiation ability [48]. Collectively, these findings above all indicate that APMs in normal adrenal glands could possibly differentiate into ZF-like cells and play important roles in development of APAs. However, at this juncture, the transition from APMs to APAs remains unestablished.
POTENTIAL ASSOCIATION OF APM AND APA
- The diagnosis of overt PA is generally based upon the value of the aldosterone-to-renin ratio in severe or resistant hypertension. Overt PA is mostly composed of APA and BHA and it is clinically not difficult to differentiate between these two entities. However, it is also true that some individuals have normotension and normal electrolyte values accompanied by suppressed plasma renin activity with normal or slightly elevated plasma aldosterone levels [49]. These subjects demonstrated a tendency for renin-independent aldosteronism, which could place them at a higher risk of developing clinically meaningful hypertension [49]. Therefore, we tentatively termed this condition as subclinical PA [49]. One of the potentially frequent causes of subclinical PA is considered the presence of APMs in the normal adrenal cortex [11,37]. APMs frequently harbor APA-driver somatic mutations and the number of mutated APMs was found to increase with aging [11,37]. We also demonstrated the presence of APMs in normal adrenal gland adjacent to APAs with autonomous aldosterone production [37]. These findings above all indicate that the development of APMs in the normal adrenal glands is indeed independent of the status of RAAS in subjects (Fig. 3). This formation of RAAS-independent APMs and APA or APN is therefore reasonably postulated to result from aging, but further investigations are warranted for clarification.
SUBCLINICAL OR NORMOTENSIVE PA
- Aging is a natural process characterized by a progressive loss of physiological function, which is especially marked in the endocrine system. Many earlier studies demonstrated an age-dependent decline in the activity of the RAAS in normal human subjects regardless the status of sodium repletion [50,51]. This decline in the RAAS with increased aging is generally considered to result from decreased plasma renin activity [52]. One possible effect of this phenomenon may be the deterioration of renal function with aging [53]. In addition, the response of aldosterone to angiotensin II (ANGII) was more sensitive in younger adult rats, but decreased in older ones [54]. This was further confirmed in human female subjects, but of particular interest, not in males [55]. The progressive decrement of plasma renin activity despite unchanged plasma and urinary aldosterone levels was reported in elderly subjects without concomitant evidence of PA in the supine position [56]. Physiological RAAS-independent autonomous aldosterone production was proposed to develop possibly due to changes of ZG with aging [56].
- The age-dependent decline of aldosterone levels is not only due to decreased renin or ANGII levels, but also due to age-associated structural changes. For instance, the ZG becomes atrophic in older people compared with young adults [57,58]. The number of APMs in the normal ZG was also reported to increase during aging [11,56]. However, with the increased number of APMs, the total CYP11B2-expressing area in APMs was significantly negatively correlated with aging [56]. Based on those results, RAAS-independent development of APMs with aging leads to more autonomous aldosterone production, but less physiological aldosterone production [56]. In addition, adrenocortical nodule formation is well known to increase with aging by classical autopsy studies [57]. Moreover, the ZG cells lost in elderly people were replaced by abundant progenitor cells, which implied the potential differentiation to APMs or APA [58]. Collectively, aging is a risk factor in the transformation of “physiological” to “pathological” adrenocortical nodules and possibly to APAs, which all could contribute to autonomous aldosterone production. This autonomous aldosterone production leads to a potentially high risk of incident hypertension in elder people.
AGING AND ALDOSTERONE
- Aldosterone-producing disorders are the major cause of PA or normotensive PA with autonomous aldosterone secretion. These disorders frequently harbor somatic mutations in ion channels or pumps including KCNJ5, CACNA1D, ATP1A1, and ATP2B3. Somatic mutations may induce a secondary differentiation of adrenocortical cells into compact or clear cells. All of these contribute to normotensive PA and overt PA depending on the subtype of the disorder (APMs, APNs, and APAs). The formation of these disorders remains unclear, but may possibly be due to somatic mutations or aging, which converts physiological adrenocortical morphology to pathological disorders. In addition, these disorders in elder people secrete RAAS-independent aldosterone, which accounts for the high risk of incident hypertension or other cardiovascular events.
CONCLUSIONS
-
Acknowledgements
- Fumitoshi Satoh and Hironobu Sasano were supported by grants from Ministry of Health, Labour and Welfare in Japan (H 29 Nanji-Japan 046).
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information


- 1. Funder JW. Medicine: the genetics of primary aldosteronism. Science 2011;331:685–6.ArticlePubMed
- 2. Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006;48:2293–300.ArticlePubMed
- 3. Williams JS, Williams GH, Raji A, Jeunemaitre X, Brown NJ, Hopkins PN, et al. Prevalence of primary hyperaldosteronism in mild to moderate hypertension without hypokalaemia. J Hum Hypertens 2006;20:129–36.ArticlePubMed
- 4. Byrd JB, Turcu AF, Auchus RJ. Primary aldosteronism: practical approach to diagnosis and management. Circulation 2018;138:823–35.ArticlePubMedPMC
- 5. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab 2010;95:2296–305.ArticlePubMed
- 6. Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, et al. International histopathology consensus for unilateral primary aldosteronism. J Clin Endocrinol Metab 2021;106:42–54.ArticlePubMed
- 7. Young WF, Stanson AW, Thompson GB, Grant CS, Farley DR, van Heerden JA. Role for adrenal venous sampling in primary aldosteronism. Surgery 2004;136:1227–35.ArticlePubMed
- 8. Toniato A, Bernante P, Rossi GP, Pelizzo MR. The role of adrenal venous sampling in the surgical management of primary aldosteronism. World J Surg 2006;30:624–7.ArticlePubMed
- 9. Omata K, Satoh F, Morimoto R, Ito S, Yamazaki Y, Nakamura Y, et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension 2018;72:874–80.ArticlePubMedPMC
- 10. Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, et al. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J Clin Endocrinol Metab 2017;102:1182–92.ArticlePubMed
- 11. Omata K, Anand SK, Hovelson DH, Liu CJ, Yamazaki Y, Nakamura Y, et al. Aldosterone-producing cell clusters frequently harbor somatic mutations and accumulate with age in normal adrenals. J Endocr Soc 2017;1:787–99.ArticlePubMedPMC
- 12. Nishimoto K, Seki T, Kurihara I, Yokota K, Omura M, Nishikawa T, et al. Case report: nodule development from subcapsular aldosterone-producing cell clusters causes hyperaldosteronism. J Clin Endocrinol Metab 2016;101:6–9.ArticlePubMed
- 13. Hegstad R, Brown RD, Jiang NS, Kao P, Weinshilboum RM, Strong C, et al. Aging and aldosterone. Am J Med 1983;74:442–8.ArticlePubMed
- 14. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011;331:768–72.ArticlePubMedPMC
- 15. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 2013;45:1055–60.ArticlePubMed
- 16. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013;45:1050–4.ArticlePubMedPMC
- 17. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013;45:440–4.ArticlePubMed
- 18. Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 2016;6:19546.ArticlePubMedPMC
- 19. Dutta RK, Arnesen T, Heie A, Walz M, Alesina P, Soderkvist P, et al. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur J Endocrinol 2019;181:K37–41.ArticlePubMed
- 20. Cheng CJ, Sung CC, Wu ST, Lin YC, Sytwu HK, Huang CL, et al. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J Clin Endocrinol Metab 2015;100:E155–63.ArticlePubMed
- 21. Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, Kikuchi K, et al. Voltage-gated calcium channels in the human adrenal and primary aldosteronism. J Steroid Biochem Mol Biol 2014;144(Pt B):410–6.ArticlePubMed
- 22. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014;64:354–61.ArticlePubMed
- 23. Di Leva F, Domi T, Fedrizzi L, Lim D, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch Biochem Biophys 2008;476:65–74.ArticlePubMed
- 24. Bonnet-Serrano F, Bertherat J. Genetics of tumors of the adrenal cortex. Endocr Relat Cancer 2018;25:R131–52.ArticlePubMed
- 25. Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015;4:e06315.ArticlePubMedPMC
- 26. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 2016;13:225–36.ArticlePubMedPMC
- 27. Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M, et al. Role for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronism. Hypertension 2014;63:783–9.ArticlePubMed
- 28. Korah HE, Scholl UI. An update on familial hyperaldosteronism. Horm Metab Res 2015;47:941–6.ArticlePubMed
- 29. Stowasser M, Bachmann AW, Huggard PR, Rossetti TR, Gordon RD. Severity of hypertension in familial hyperaldosteronism type I: relationship to gender and degree of biochemical disturbance. J Clin Endocrinol Metab 2000;85:2160–6.ArticlePubMed
- 30. Lifton RP, Dluhy RG, Powers M, Ulick S, Lalouel JM. The molecular basis of glucocorticoid-remediable aldosteronism, a Mendelian cause of human hypertension. Trans Assoc Am Physicians 1992;105:64–71.PubMed
- 31. Stowasser M, Wolley M, Wu A, Gordon RD, Schewe J, Stolting G, et al. Pathogenesis of familial hyperaldosteronism type II: new concepts involving anion channels. Curr Hypertens Rep 2019;21:31.ArticlePubMed
- 32. Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 2018;50:355–61.ArticlePubMed
- 33. Nanba K, Omata K, Else T, Beck PC, Nanba AT, Turcu AF, et al. Targeted molecular characterization of aldosterone-producing adenomas in White Americans. J Clin Endocrinol Metab 2018;103:3869–76.ArticlePubMedPMC
- 34. De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, Rainey WE, et al. Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension 2020;75:1034–44.ArticlePubMedPMC
- 35. Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J Atheroscler Thromb 2015;22:191–200.ArticlePubMed
- 36. Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, et al. Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J Clin Endocrinol Metab 2013;98:1567–74.ArticlePubMed
- 37. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A 2015;112:E4591–9.ArticlePubMedPMC
- 38. Yamazaki Y, Omata K, Tezuka Y, Ono Y, Morimoto R, Adachi Y, et al. Tumor cell subtypes based on the intracellular hormonal activity in KCNJ5-mutated aldosterone-producing adenoma. Hypertension 2018;72:632–40.ArticlePubMed
- 39. Gao X, Yamazaki Y, Tezuka Y, Pieroni J, Ishii K, Atsumi N, et al. Intratumoral heterogeneity of the tumor cells based on in situ cortisol excess in cortisol-producing adenomas: an association among morphometry, genotype and cellular senescence. J Steroid Biochem Mol Biol 2020;204:105764.ArticlePubMed
- 40. Neville AM, O’Hare MJ. The human adrenal cortex. Pathology and biology: an integrated approach; Berlin: Springer; 1982.Article
- 41. Tsuchiyama H, Kawai K, Harada T, Shigematsu K, Sugihara H. Functional pathology of aldosterone-producing adenoma. Acta Pathol Jpn 1980;30:967–76.ArticlePubMed
- 42. Ono Y, Yamazaki Y, Omata K, Else T, Tomlins SA, Rhayem Y, et al. Histological characterization of aldosterone-producing adrenocortical adenomas with different somatic mutations. J Clin Endocrinol Metab 2020;105:e282–9.ArticlePubMed
- 43. Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, et al. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab 2012;97:E819–29.ArticlePubMed
- 44. Gioco F, Seccia TM, Gomez-Sanchez EP, Rossi GP, Gomez-Sanchez CE. Adrenal histopathology in primary aldosteronism: is it time for a change? Hypertension 2015;66:724–30.ArticlePubMedPMC
- 45. Boulkroun S, Samson-Couterie B, Dzib JF, Lefebvre H, Louiset E, Amar L, et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension 2010;56:885–92.ArticlePubMed
- 46. Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology 2011;152:4753–63.ArticlePubMed
- 47. Hammer GD, Basham KJ. Stem cell function and plasticity in the normal physiology of the adrenal cortex. Mol Cell Endocrinol 2021;519:111043.ArticlePubMed
- 48. Freedman BD, Kempna PB, Carlone DL, Shah M, Guagliardo NA, Barrett PQ, et al. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell 2013;26:666–73.ArticlePubMedPMC
- 49. Brown JM, Robinson-Cohen C, Luque-Fernandez MA, Allison MA, Baudrand R, Ix JH, et al. The spectrum of subclinical primary aldosteronism and incident hypertension: a cohort study. Ann Intern Med 2017;167:630–41.ArticlePubMedPMC
- 50. Weidmann P, De Myttenaere-Bursztein S, Maxwell MH, de Lima J. Effect on aging on plasma renin and aldosterone in normal man. Kidney Int 1975;8:325–33.PubMed
- 51. Noth RH, Lassman MN, Tan SY, Fernandez-Cruz A Jr, Mulrow PJ. Age and the renin-aldosterone system. Arch Intern Med 1977;137:1414–7.ArticlePubMed
- 52. Tsunoda K, Abe K, Goto T, Yasujima M, Sato M, Omata K, et al. Effect of age on the renin-angiotensin-aldosterone system in normal subjects: simultaneous measurement of active and inactive renin, renin substrate, and aldosterone in plasma. J Clin Endocrinol Metab 1986;62:384–9.ArticlePubMed
- 53. Laragh JH, Sealey JE. The plasma renin test reveals the contribution of body sodium-volume content (V) and renin-angiotensin (R) vasoconstriction to long-term blood pressure. Am J Hypertens 2011;24:1164–80.ArticlePubMed
- 54. Rakotondrazafy J, Brudieux R. Age-related change in plasma aldosterone response to exogenous angiotensin II in the rat. Horm Res 1993;39:156–60.ArticlePubMed
- 55. Giacche M, Vuagnat A, Hunt SC, Hopkins PN, Fisher ND, Azizi M, et al. Aldosterone stimulation by angiotensin II: influence of gender, plasma renin, and familial resemblance. Hypertension 2000;35:710–6.ArticlePubMed
- 56. Nanba K, Vaidya A, Williams GH, Zheng I, Else T, Rainey WE. Age-related autonomous aldosteronism. Circulation 2017;136:347–55.ArticlePubMedPMC
- 57. Hornsby PJ. Aging of the human adrenal cortex. Ageing Res Rev 2002;1:229–42.ArticlePubMed
- 58. Aiba M, Fujibayashi M. Alteration of subcapsular adrenocortical zonation in humans with aging: the progenitor zone predominates over the previously well-developed zona glomerulosa after 40 years of age. J Histochem Cytochem 2011;59:557–64.ArticlePubMedPMCPDF
References
Figure & Data
References
Citations
- Subtype-specific Body Composition and Metabolic Risk in Patients With Primary Aldosteronism
Seung Shin Park, Chang Ho Ahn, Sang Wan Kim, Ji Won Yoon, Jung Hee Kim
The Journal of Clinical Endocrinology & Metabolism.2024; 109(2): e788. CrossRef - 2023 Korean Endocrine Society Consensus Guidelines for the Diagnosis and Management of Primary Aldosteronism
Jeonghoon Ha, Jung Hwan Park, Kyoung Jin Kim, Jung Hee Kim, Kyong Yeun Jung, Jeongmin Lee, Jong Han Choi, Seung Hun Lee, Namki Hong, Jung Soo Lim, Byung Kwan Park, Jung-Han Kim, Kyeong Cheon Jung, Jooyoung Cho, Mi-kyung Kim, Choon Hee Chung
Endocrinology and Metabolism.2023; 38(6): 597. CrossRef - Correlation of Histopathologic Subtypes of Primary Aldosteronism with Clinical Phenotypes and Postsurgical Outcomes
Chang Ho Ahn, You-Bin Lee, Jae Hyeon Kim, Young Lyun Oh, Jung Hee Kim, Kyeong Cheon Jung
The Journal of Clinical Endocrinology & Metabolism.2023;[Epub] CrossRef - Expression of CYP11B1 and CYP11B2 in adrenal adenoma correlates with clinical characteristics of primary aldosteronism
Chang Ho Ahn, Hee Young Na, So Yeon Park, Hyeong Won Yu, Su‐Jin Kim, June Young Choi, Kyu Eun Lee, Sang Wan Kim, Kyeong Cheon Jung, Jung Hee Kim
Clinical Endocrinology.2022; 96(1): 30. CrossRef - Pathology of Aldosterone Biosynthesis and its Action
Xin Gao, Yuto Yamazaki, Yuta Tezuka, Kei Omata, Yoshikiyo Ono, Ryo Morimoto, Yasuhiro Nakamura, Takashi Suzuki, Fumitoshi Satoh, Hironobu Sasano
The Tohoku Journal of Experimental Medicine.2021; 254(1): 1. CrossRef - Cellular Senescence in Adrenocortical Biology and Its Disorders
Xin Gao, Faping Li, Bin Liu, Yuxiong Wang, Yishu Wang, Honglan Zhou
Cells.2021; 10(12): 3474. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite



