Sarcopenia and Muscle Aging: A Brief Overview
Article information

Abstract
The world is facing the new challenges of an aging population, and understanding the process of aging has therefore become one of the most important global concerns. Sarcopenia is a condition which is defined by the gradual loss of skeletal muscle mass and function with age. In research and clinical practice, sarcopenia is recognized as a component of geriatric disease and is a current target for drug development. In this review we define this condition and provide an overview of current therapeutic approaches. We further highlight recent findings that describe key pathophysiological phenotypes of this condition, including alterations in muscle fiber types, mitochondrial function, nicotinamide adenine dinucleotide (NAD+) metabolism, myokines, and gut microbiota, in aged muscle compared to young muscle or healthy aged muscle. The last part of this review examines new therapeutic avenues for promising treatment targets. There is still no accepted therapy for sarcopenia in humans. Here we provide a brief review of the current state of research derived from various mouse models or human samples that provide novel routes for the development of effective therapeutics to maintain muscle health during aging.
INTRODUCTION
Worldwide advances in applied science and health care technology, along with socioeconomic development, has increased lifespan. In 2019, it was estimated that globally 703 million people were older than 65 years old and that this number would reach 1.5 billion by 2050 [1]. All nations have seen an increase in life expectancy with a disproportionately greater increase in elderly populations [1]. Consequently, healthcare services are strained under the additional burden of increasing elderly populations. A better understanding of age-related chronic diseases and new modalities are required to address aging-related diseases and alleviate pressure on healthcare systems.
One such disease is sarcopenia. Sarcopenia is the progressive and generalized loss of muscle mass and function (i.e., strength) that occurs with age [2] and is strongly associated with frailty. Frailty is defined by the vulnerability of the elderly to possible stressors that increase risk for poor health outcomes, incident disability, and mortality [3]. Recent studies have revealed that sarcopenia is not a simple condition caused by nutrient deficiency or a sedentary lifestyle, but a result of an elaborate multi-pathway pathogenesis [4,5]. This article presents a brief overview of the current knowledge of the underlying mechanisms of sarcopenia along with novel therapies being explored to prevent and treat this condition.
SARCOPENIA
Sarcopenia
The Aging in Motion (AIM) Coalition describes sarcopenia as a type of persistent muscle atrophy characterized by gradual loss of skeletal muscle mass and function (strength) with a risk of negative outcomes such as physical disability, poor quality of life and death. Additionally, in 2016, sarcopenia became officially classified as a disease state under the International Classification of Diseases, Clinical Modification (ICD-10-CM) code (M62.84) [6], thus emphasizing the need for improved diagnostic methods and drug development to treat sarcopenia. For instance, in 1998 using dual X-ray absorptiometry, Baumgartner et al. [7] proposed sarcopenia as any appendicular skeletal muscle mass (kg)/height (m2) that was >2 standard deviations (SDs) beneath that of a young reference population’s mean. Similarly, in 2003, using computed tomography scans of calf muscle, Lauretani et al. [8] defined sarcopenia as a muscle cross-sectional area that was >2 SDs below the population mean. In 2010, the European Working Group on Sarcopenia in Older People (EWGSOP) suggested that sarcopenia should be clinically defined by both reduced muscle quantity and degraded muscle quality (i.e., strength or performance) [9]. Eight years later, based on more laboratory and clinical evidence, EWGSOP updated their consensus. They determined that low muscle strength was the primary defining attribute of sarcopenia, and that low muscle mass and quality are confirmatory markers of sarcopenia diagnosis, with severe sarcopenia defined by additional impaired physical performance [10].
Currently, many regional and international organizations, such as the EWGSOP, the Asian Working Group for Sarcopenia (AWGS), the International Working Group on Sarcopenia (IWGS), and the Foundation for the National Institutes of Health (FNIH), have proposed various diagnostic criteria and screening stratgeis for sarcopenia, which are summarized in Table 1 [10–13]. In general, these groups agree on diagnosing sarcopenia using both muscle mass and muscle function together; however, they use different measurement tools and thresholds. Despite these differences, sarcopenia is generally diagnosed in about 20% of elderly research subjects with similar proportions for Caucasian and Asian populations [14,15]. Additionally, sarcopenia is exceptionally common in people with cardiovascular disease, dementia, diabetes mellitus, and respiratory disorders [16].

Summary of Recommendations in the Diagnostic Criteria for Sarcopenia
AGING OF SKELETAL MUSCLE
Anabolic resistance
Normally, muscle contraction, dietary amino acids, and anabolic hormones, such as insulin like growth factor-1 (IGF-1), are potent stimulators of muscle growth. However, when muscle becomes resistant to these stimuli this is termed anabolic resistance (AR). AR is a phenomenon defined by the diminished ability to increase the rate of protein synthesis and adapt to anabolic stimulation [17,18]. AR is often observed in aged muscle and is probably a crucial driver of sarcopenia and physical frailty [19,20]. It may underlie the greater requirement for dietary protein in older adults [21]. Although the underlying mechanisms remain to be fully elucidated, it is primarily thought to be due to reduced sensitivity of AKT (also known as protein kinase B [PKB])-mammalian target of rapamycin (mTOR) kinase cascade [17,19,21,22]. AR is likely one of many contributing factors to sarcopenia.
Muscle fiber-type
Muscle consists of force-generating contractile muscle fibers bundled together by connective tissue. The sarcomere is the functional unit of muscle fiber, containing thin filaments (including actin, tropomyosin, and troponin) and thick filaments (primarily comprised of myosin), along with other structural proteins. ATP-dependent crawling of myosin filaments along actin chains generates muscle contraction. Mature myosin protein contains six polypeptides, four light chains, and two heavy chains [23]. The expression of different myosin heavy chain (MHC) isoforms determines muscle fiber types. Classically, adult human skeletal myofibers have been classified as either slow-twitch (type I) or fast-twitch (type II) fiber types, according to their contractile speed in response to neural excitement. Presently, myofibers are classified as type I, type IIA, IIB, or IIX, in parallel with the MHC isoform expressed. Type I and IIA fibers predominantly depend on mitochondrial oxidative phosphorylation for energy production, whereas type IIB and IIX rely on glycolysis [23].
Several reports show that aging causes muscle fiber-type transition that is characterized by a gradual decrease of both type II fiber number and size with age [24,25] suggesting a fast to slow fiber-type shift. This is further supported by a study that found that expression of IIA and IIX MHC mRNA decreased by 14% and 10%, respectively, per decade while type I mRNA remained constant with age [26]. Additionally, by analyzing single myofiber expression of MHC proteins, Klitgaard et al. [27] found that with age there is an increase in fibers co-expressing multiple MHC isoforms. Increased clustering of myofibers of similar types has been shown to be associated with both samples from patients with spinal muscular atrophy and in subjects over the age of 50 [28]. This muscle fiber-type shift from fast to slow may also suggest that with aging skeletal muscle transitions from relying primarily on cytosolic glycolytic metabolism to oxidative metabolism and thus has a greater requirement for functional mitochondria.
Mitochondrial dysfunction
Although, classically, mitochondria are considered the cellular power plants of eukaryotic cells, recent evidence describes mitochondria as multifunctional signaling hubs, linking various cellular functions to metabolic and age-associated diseases [29]. Mitochondria play pivotal roles in cellular homeostasis responding to intracellular and extracellular stresses, cell death, inflammation, epigenetics, and senescence [30]. Mitochondrial dysfunction describes the functional decline of mitochondrial quality and activity due to destruction of mitochondrial structure, reduction of mitochondrial content, malfunction of mitochondrial metabolism and oxidative phosphorylation, accumulation of mitochondrial DNA damage, and dysregulation of mitochondrial dynamics (fusion and fission) [31] and is a well-recognized “hallmark of aging” [29,32].
Many reports have demonstrated that sarcopenia is tightly associated with mitochondrial dysfunction in skeletal muscles as well as motor neurons [33,34]. For instance, recently, using muscle biopsies from 119 human elderly subjects drawn from Caucasian, African, and Asian cohorts, Migliavacca et al. [35] found reduced mitochondrial oxidative capacity and nicotinamide adenine dinucleotide (NAD+) biosynthesis in sarcopenic individuals. Furthermore, several studies have proposed that mitochondrial dysfunction in motor neurons drives sarcopenia [36,37]. In addition, studies using muscle transcriptomes revealed reduced expression of many mitochondrial genes, including peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) and estrogen related receptors (ERRs), both key regulatory factors of mitochondrial biogenesis and homeostasis [38]. In our unpublished work analyzing publicly available Genotype-Tissue Expression (GTEx) data and National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) data, the expression of the majority of both nuclear and mitochondrial DNA-encoded mitochondrial genes is reduced in muscle from aged rodents and humans.
Interestingly, mitophagy, which selectively eliminates damaged or dysfunctional mitochondria via autophagy, is also reduced during normal aging [39]. The requirement of autophagy for muscle maintenance was demonstrated with the observation that mice lacking Atg7 in muscle, a key enzyme required for muscle autophagy, experience premature muscle dysfunction and sarcopenia [40]. Similarly, overexpression of Atg5 in mice increases autophagic protein expression and prevents age-induced decreases in muscle function [41]. This finding suggests that autophagy may be required to maintain muscle function with age. Several recent studies demonstrated that the enhancement of mitophagy by the administration of single compounds, such as urolithins [42–44] and spermidine [45], led to beneficial effects in aged skeletal muscle, heart, and neurons. These data suggest that mitophagy enhancers could provide new therapeutic options to treat sarcopenia. In summary, mitochondrial dysfunction is associated with sarcopenia with many contributing factors. These include but are not limited to reduced PGC-1α and ERR signaling, insufficient autophagy/mitophagy, and reduced muscular NAD+ levels with age.
NAD+ metabolism
NAD+ is a well-known essential biomolecule. It is a substrate for many enzymes, including poly(ADP-ribose) polymerase 1 (PARP1) and sirtuin 1 (SIRT1), and essential for metabolic processes where it accepts and donates electrons for glycolysis, β-oxidation, the tricarboxylic acid cycle, and the electron transport chain [46]. Numerous independent studies have found that cellular NAD+ levels decline in the majority of tissues, including skeletal muscle, liver, adipose tissue, and adult stem cells, with aging and pathological conditions [47–49]. This suggests that NAD+ is associated with aging and presumably diseases such as sarcopenia. As aforementioned, NAD+ biosynthesis is reduced in skeletal muscle from Caucasian, African, and Asian sarcopenic cohorts [35]. In support of a causative role of NAD+ deficiency in muscular atrophy, mice with impaired NAD+ salvage pathways in muscle, i.e., skeletal muscle-specific NAMPT knockout (KO) mice, Frederick et al. [49] observed increased mitochondrial dysfunction, decreased muscle mass and function, that was exacerbated with age. Subsequently, researchers investigated the therapeutic potential of NAD+ boosting molecules [50]. NAD+ replenishment using NAD+ boosters, including nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), nicotinamide (NAM), CD38 inhibitors and PARP inhibitors, protects from age- or congenital disease-related muscle phenotypes [51–55]. NR supplementation alone improves muscle satellite cell regenerative capacity and rescues the muscular atrophy phenotype of mice lacking skeletal muscle NAMPT [49]. Similarly, overexpression of a constitutively active NAMPT maintains muscle NAD+ levels and exercise performance in old mice, demonstrating the essential role of tissue-autonomous NAD+ homeostasis [55,56].
MYOKINES AND MUSCLE AGING
Myostatin and follistatin
Myostatin (encoded by the MSTN gene, also known as growth differentiation factor 8 [GDF-8]) is a myokine that negatively regulates myogenesis [57]. It belongs to the transforming growth factor-β (TGFβ) family, is secreted from muscle, and has local (autocrine) or systemic (endocrine) effects by acting on activin type II A and B (ActRIIA/B) receptors. Animal models lacking myostatin and humans with congenital myostatin loss experience pronounced muscle loss and indicated the negative relationship between myostatin expression and muscle size [58–60]. In contrast, follistatin, also a member of the TGFβ family, is a competitive antagonist for myostatin binding to the ActRIIB receptor [57,58]. Follistatin was first found in ovarian follicular fluid and later in skeletal muscle, testis, liver and many different tissues [57,58]. Reciprocal to ablation of myostatin, overexpression or administration of recombinant follistatin causes muscle hypertrophy [57,58,61]. Thus, myostatin and follistatin are both known regulators of muscle mass.
However, the role of myostatin and follistatin in sarcopenia is unclear. In C2C12 myoblasts, myostatin was elevated in late passage (>30 passages) versus early passage (<12 passages) [62]. These findings have been confirmed by some studies in older humans and animal subjects where with increasing age and sarcopenia there is also increased expression of myostatin [63,64]. However, not all studies come to the same conclusion. Bergen et al. [63] observed this to be true only in female but not male participants, supporting the increased incidence of sarcopenia in females. Furthermore, Ratkevicius et al. [65] observed no differences in serum myostatin between young and sarcopenic elderly men. Similarly, follistatin does not appear to change with age [63,66]. These conflicting findings suggest that despite their clear potential for regulation of muscle mass myostatin and follistatin may not directly contribute to sarcopenia or muscle aging.
GDF11 and GDF15
Growth differentiation factors (GDFs), including GDF11 and GDF15, belong to the TGFβ protein superfamily. They share homeostatic and metabolic regulation functions with TGFβ-family members (such as myostatin) and bone morphogenic proteins (BMPs). Furthermore, they are both associated with regulation of skeletal muscle function and aging.
GDF11 has an analogical molecular structure and similar signaling pathways to myostatin [67]. However, GDF11 has different tissue distribution and biological effects. According to the GTEx database [68], GDF11 is broadly expressed in various tissues, including the brain, spleen, female reproductive organs, and digestive tract, while myostatin is predominantly expressed in the cervix, cerebellum, adrenal gland, skeletal muscle, aorta, and pituitary. Whereas myostatin’s effects on skeletal muscle are well established, the perinatal lethality of GDF11 KO mice has restricted our knowledge of GDF11’s role in skeletal muscle aging.
The current literature presents conflicting findings for the role of GDF11 in aging-related skeletal muscle phenotypes. GDF11 protein levels are higher in muscles from young mice compared to aged mice [69]. Further, Sinha et al. [69] found that administering recombinant GDF11 to aged mice reduced aging-associated DNA damage, improved mitochondrial and myofibrillar morphology, increased neuromuscular junction (NMJ) formation, improved endurance and grip strength, and improved muscle regeneration. However, Egerman et al. [70] reported conflicting results. After finding that the previously used reagents for GDF11 measurement were not specific and in contrast to the previous work, they found a trend for elevated circulating GDF11 in aged rats and older humans. Additionally, they found that GDF11 induced similar gene changes, signaling and inhibition of myoblast differentiation as myostatin. Finally, they demonstrated that GDF11 inhibits rather than helps muscle regeneration [71,70]. This has been further confirmed by other laboratories that have shown skeletal muscle atrophy [72] and impaired skeletal muscle regeneration [73] following recombinant GDF11 administration. Furthermore, administration of a GDF11/myostatin inhibitory peptide (GDF11PRO-Fc) induces skeletal muscle hypertrophy [74].
GDF15, also known as macrophage inhibitory cytokine 1 (MIC-1), traditionally has been categorized as a member of the TGFβ superfamily and has also been investigated for its effects on skeletal muscle. Recent studies have identified glial cell line-derived neurotrophic factor (GDNF) family receptor alpha like (GFRAL) as a receptor for GDF15 and thus raised the possibility that GDF15 shares similar actions to the GDNF-like growth factor family [75,76]. The Shong group proposed that GDF15 is a myomitokine, released in response to activation of the mitochondrial unfolded protein response, that beneficially regulates systemic energy expenditure [77,78]. Moon et al. [79] recently reported that Mendelian randomization analysis showed a causal effect of reduced GDF15 levels in human blood samples resulting in increased body weight and inflammation. The administration of recombinant GDF15 protein has an anti-obesity effect in mice [80,81]. In contrast to studies proposing beneficial effects of GDF15 on body mass, Lerner et al. [82] reported that GDF15 is a cytokine secreted from tumors that drives cancer cachexia. Furthermore, administration of GDF15 neutralizing antibodies prevents cancer and GDF-11-induced cachexia [83]. Consistent with this, Suriben et al. [84] found an inhibitory antibody raised against the GFRAL extracellular domain also prevented tumor-induced weight loss. However, most of these studies show the weight loss to be primarily driven by a loss in adipose tissue, consistent with the low expression of GFRAL in skeletal muscle (as measured in the GTEx database). Although GDF15 has been shown to be released from muscle and generates a negative caloric balance, more studies are required to test if it plays a causative role in muscular atrophy or sarcopenia.
GUT MICROBIOTA AND MUSCLE AGING
Recent studies reported that the diversity and composition of gut microbiome—the entire set of microorganisms residing in the digestive system of the host—could be altered significantly with aging [85,86]. Age-associated alterations of the gut microbiome might be related to decreased anabolic stimuli-induced muscle protein synthesis (i.e., AR) and chronic inflammation, which affects all aspects of skeletal muscle, including metabolism and physical performance [87–89]. The concept of an aging-related “leaky gut,” whereby elderly hosts have abnormally increased gut mucosa permeability, suggests that the pro-inflammatory signals from the gut microbiome can enter the bloodstream and cause chronic low grade inflammation in the elderly [90]. If true, this may also be an avenue by which the age-associated changes in the gut microbiome may affect skeletal muscle.
Another possible candidate linking the gut-muscle axis to skeletal muscle homeostasis is gut microbiota-produced metabolites. Gut microbiota produce many metabolites, including short-chain fatty acids, that regulate host metabolism and inflammation [87]; imidazole propionate, which triggers insulin resistance [91]; indoxyl sulfate, which may be a cause of muscle atrophy [92]; and urolithin A (UA), which enhances mitophagy in skeletal muscle [42]. Thus, these findings suggest that metabolites affecting pathways thought to influence skeletal muscle homeostasis are produced by microbiota and are able to consequently influence host homeostasis independent of microbiota penetration of the intestinal lining and subsequent inflammation.
Specific studies have investigated the effect of manipulations of the gut microbiome on skeletal muscle. For example, Lahiri et al. [93] reported that germ-free mice harbor a muscular atrophy phenotype in comparison with pathogen-free mice. Germ-free mice have increased expression of muscular atrophy proteins (e.g., atrogin-1 and muscle RING finger 1 [MuRF-1]), reduced expression of IGF-1, and reduced expression of serum choline, the precursor for acetylcholine, an essential neurotransmitter in the NMJ. Administration of gut microbiota from pathogen-free mice to germ-free mice and short-chain fatty acids could improve atrophy status, including muscle mass, as well as gene expression and biomarkers [93], thus implicating the bacterial and bacterially-derived metabolites in muscular atrophy. In agreement with Lahiri et al. [93], treatment with the antibiotic metronidazole, which alters the gut microbiota by favoring recolonization with Proteobacteria and Erysipelotrichales, induced muscle atrophy in mice [94]. Interestingly, Blacher et al. [95] recently demonstrated that antibiotics eliminating gut microbiota impaired muscular function in a murine model of amyotrophic lateral sclerosis (Sod1-G93A mice) and further that gut microbiota could be a chemical factory producing favorable NAD+ precursors for host de novo NAD+ synthesis. In their study, the administration of an engineered Escherichia coli, defective in NAD+ generation, decreased grip endurance versus wild-type E. coli, whereas recolonization of antibiotic-treated mice with Akkermansia muciniphila increased levels of the NAD+ precursor, NAM, increased neuromuscular function, and extended the lifespan of Sod1-G93A mice. Additionally, two recent review articles discussed the potential connections and mechanisms between the gut microbiome and muscle aging [87,96] in more detail. In summary, the gut microbiome is a potential target for sarcopenia.
Hereby, we have covered issues related to muscle aging and sarcopenia. In summary, during the aging process, there are transitions of muscle fiber-type (a gradual decrease of both type II fiber number and size), mitochondrial dysfunction, perturbed NAD+ metabolism, and altered expressions of myokines such as myostatin, GDF11, and GDF15. Also, the composition of gut microflora and their derived metabolites, mediating the gut-muscle axis, are remodeled with age (Fig. 1).
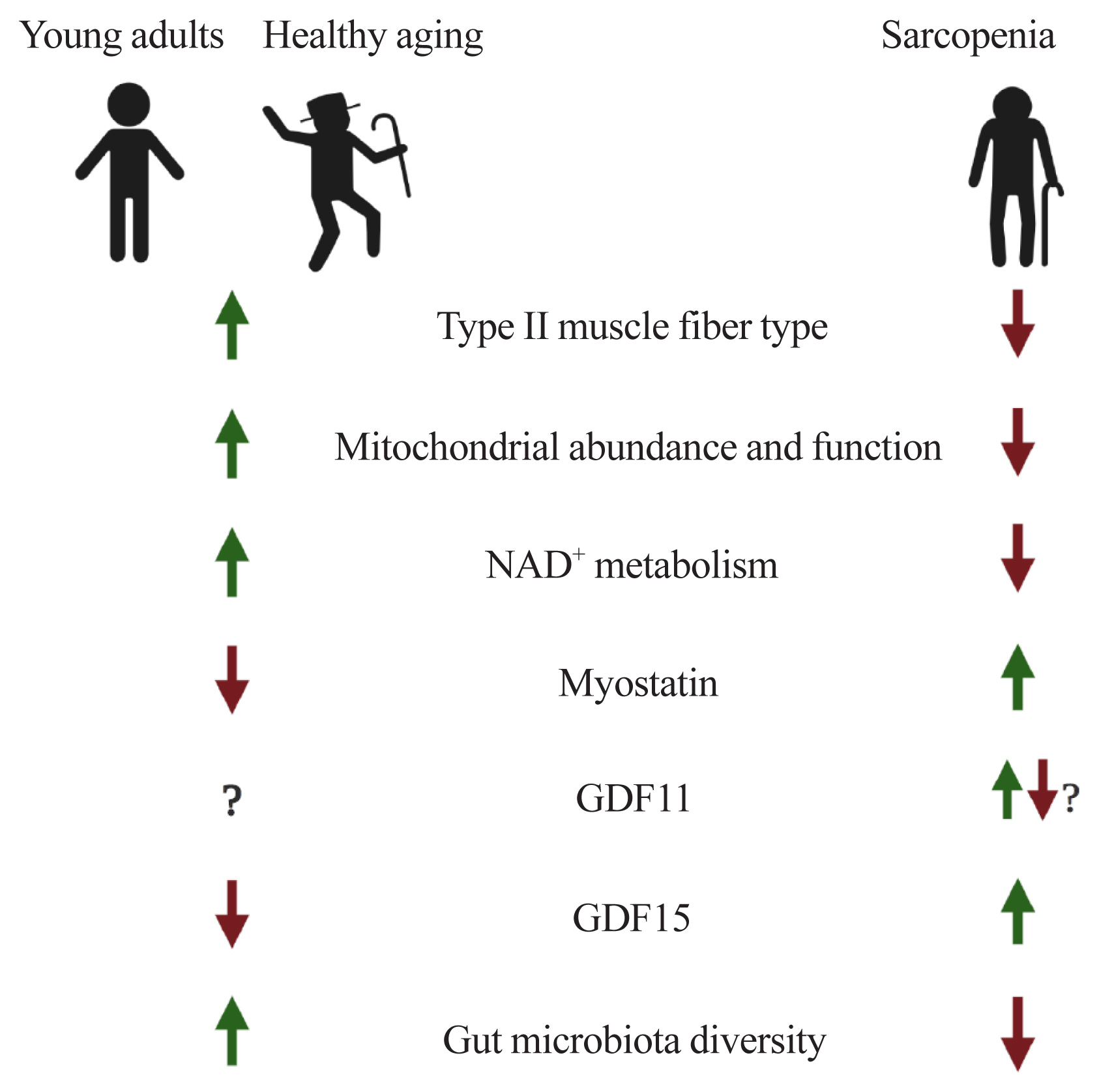
Potential factors underlying muscle aging. NAD+, nicotinamide adenine dinucleotide; GDF, growth differentiation factor.
THERAPEUTIC STRATEGIES FOR MUSCLE AGING
Anabolic resistance and amino acids
AR, as described above, is defined by a diminished muscle protein synthetic rate following anabolic stimulation such as amino acids, muscle contraction, or anabolic hormones and is often observed with aging [17,18,20,97,98]. Thus theoretically, overcoming AR by supplementing with anabolic stimulants like essential amino acids (EAA) and resistance exercise are recommended avenues to combat sarcopenia [99,100]. However, despite efforts, they have yet to yield highly effective outcomes that correct all the effects of sarcopenia [101,102]. These findings indicate that sarcopenia could be composed of several subclasses of diseases originated by distinct internal (intramuscular) or external (systemic) triggers. To overcome AR and to develop novel therapeutics for sarcopenia, multi-omics analyses including not only genome, transcriptome and proteome but also metabolome and floxome (complete set of metabolic fluxes) have to be explored [18,103].
Exercise mimetics
As mentioned above, the best therapeutic option for sarcopenia is the combination of a regular exercise program and EAA supplementation. However, some patients cannot conduct regular exercise to maintain muscle protein synthesis and mass. For instance, patients with severe sarcopenia, severe frailty, hip fracture, congenital neuromuscular diseases, and those in intensive care are not able to reap the benefits of regular exercise. To help those patients who cannot perform enough physical activity to maintain muscle mass, so-nicknamed “exercise mimetics” or “exercise in a pill” pharmaceuticals are the only available therapeutic strategy to receive some of the effects of exercise in these populations.
Although a single pharmaceutical has not been identified that can replicate all the benefits of exercise, studies have examined the benefits of activating one or multiple signaling pathways associated with exercise. One molecular target that mediates the effect of exercise is the nuclear receptor peroxisome proliferator-activated receptor delta (PPARδ, also known as PPARβ). Narkar et al. [104] demonstrated that overexpression of PPARδ and administration of the PPARδ agonist GW501516 increased expression of genes associated with mitochondrial content including Ucp3, Cpt1b, and Pdk4. When used in combination with exercise, GW50156 increased oxidative myofibers and running endurance in adult mice. However, overexpression of PPARδ alone does not reduce muscle wasting in mice in response to spinal cord transection [105]. Other reports suggest that GW501516 improves skeletal muscle mass in mouse models of muscular dystrophy [106] but has no effect on muscle mass in rats [107]. Furthermore, trials with GW501516 were discontinued due to preclinical data showing rapid induction of cancer in multiple organs [108]. Mitobridge (Boston, MA, USA) and Astellas (Tokyo, Japan) announced that their PPARδ agonist ASP0367 (also known as MA-0211), which is being developed for the treatment of primary mitochondrial myopathies, has recently been granted fast track designation by the U.S. Food and Drug Administration (FDA) (Table 2). Therefore, PPARδ agonists may prove beneficial in individuals suffering from sarcopenia through improved mitochondrial function and maintenance of oxidative type I myofibers, but may not prevent atrophy.
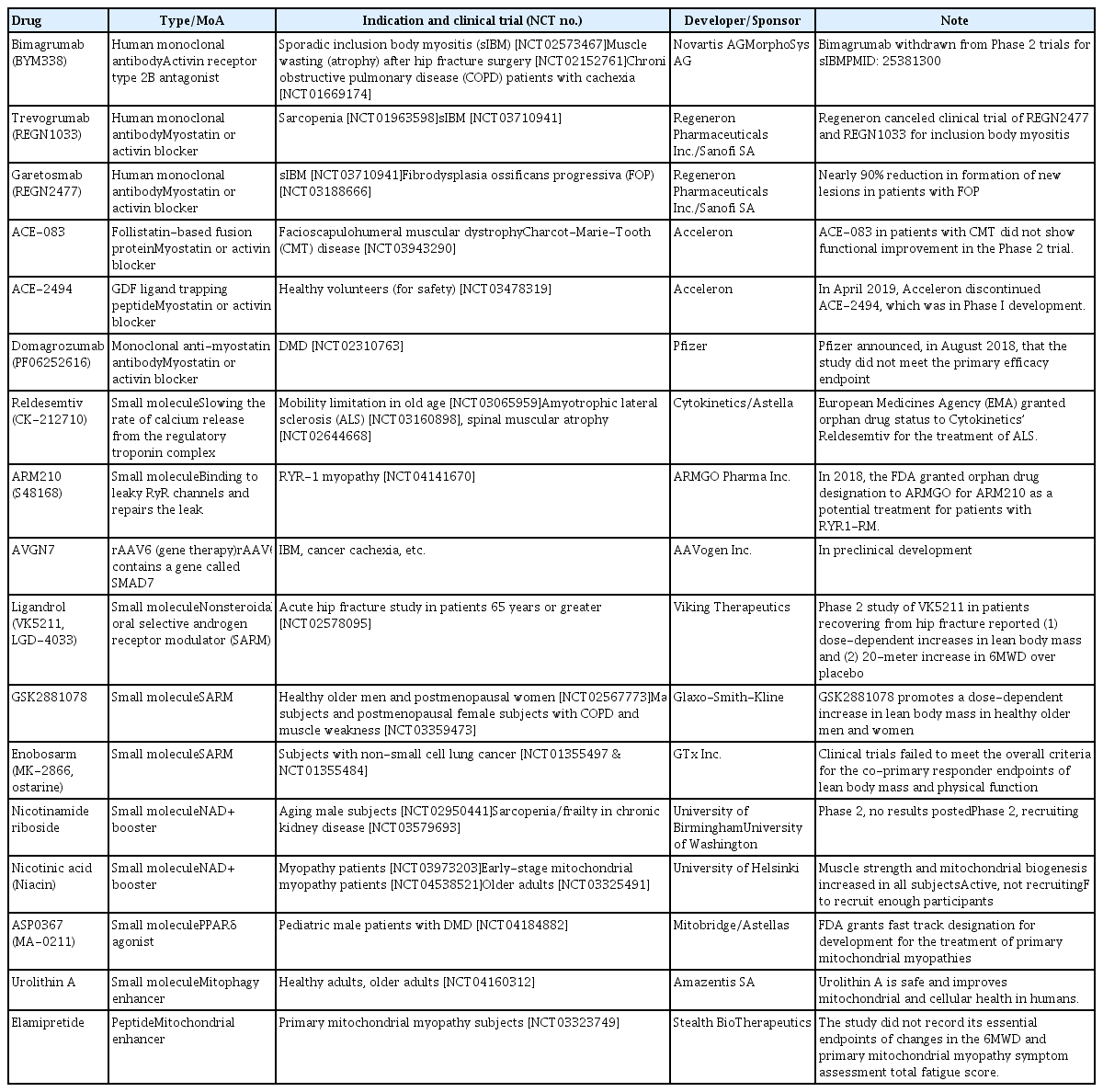
Summary of Drug Candidates Undergoing Preclinical and Clinical Trials in Muscle Wasting Diseases
Another potential therapeutic avenue for attenuating sarcopenia is pharmacological activation of the exercise activatable [109,110] and cellular energy sensor 5′-AMP-activated protein kinase (AMPK). This has been reviewed in detail elsewhere [111]. Briefly, mice with germline elimination of the AMPK structural subunit β2 and muscle-specific ablation of the structural (β1 and β2) and catalytic subunits (ɑ1 and ɑ2) of AMPK have decreased exercise capacity, decreased muscle function, and atrophied/damaged myofibers [112,113] that is exacerbated with age [114]. However, other studies have found contrasting evidence whereby expression of a dominant-negative AMPK and loss of AMPK ɑ1 and ɑ2 subunits elicited larger extensor digitorum longus muscle and larger myotubes in vitro [115,116], respectively. This may partially be explained by differences in tissue specificity of AMPK-KO models (i.e., skeletal only or heart and skeletal muscle elimination) and the conflicting regulation of skeletal myogenesis via AMPK’s known positive regulation of mitochondrial function and autophagy versus the negative regulation of protein synthesis via direct inhibition of mTOR signaling [117]. 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), which is a cell-permeable precursor to 5-amino-4-imidazole carboxamide ribonucleoside 5′-phosphate (ZMP), an allosteric activator of AMPK, increases mitochondrial content in wild-type mice [104,118] but does not increase muscle mass in wild-type or MSTN-KO mice [119]. Interestingly, it does attenuate deficits in muscle in mouse models of muscular dystrophy without affecting myofiber size [120,121] and prevents muscle loss in mouse models of cancer cachexia [122]. Furthermore, A-769662—a direct allosteric activator of AMPK β2—and AICAR synergistically attenuated interferon gamma (IFNɣ) and tumor necrosis factor-α (TNFɑ)-induced myotube atrophy in vitro. Evidence to date suggests that AMPK activators are effective at improving mitochondrial and muscle function while only maintaining skeletal muscle size in models of muscular dystrophy and cancer cachexia. New AMPK activators are continually being produced that have increased isoform specificity [123,124] and may provide further insights into tissue-specific effects of AMPK activation and their effects on maintenance or improvement of skeletal muscle. SIRT1 activators, including NAD+ boosters and resveratrol, which we describe separately, also induce exercise signaling. Absence of skeletal muscle SIRT1 reduces mitochondrial content in skeletal muscle [125]. The Auwerx group and others have demonstrated that resveratrol supplementation increases mitochondrial biogenesis, energy expenditure, and endurance exercise in mice [126]. Furthermore, it improves mitochondrial function and lipid and glucose homeostasis in obese men [127]. These results suggest that SIRT1 activation may stimulate mitochondrial content and presumably age-related decreases in skeletal muscle mitochondrial function.
In addition to the above pathways, transcription factors such as PPARα/δ, ERRα/γ, and nuclear respiratory factor-1/-2 (NRF-1/-2) contribute to the control of nuclear DNA-encoded mitochondrial gene expression [38,128], and thus any agonist or activator targeting those transcription factors may induce exercise-like effects. Although the potential of ERRγ agonists has not yet been validated in vivo, GSK4716, an ERRγ agonist, robustly upregulates genes involved in mitochondrial biogenesis and metabolism [129]. Interestingly, a recent human study administered UA for 4 weeks and showed that the muscle transcriptome of UA-given elderly individuals demonstrated a positively enriched mitochondrial gene-set profile compared to the placebo control that is similar to transcriptome profiles of muscle from active versus sedentary individuals [43]. Although exercise mimetics cannot produce all the beneficial effects of regular exercise and there is also a risk of their abuse by athletes, mimetics may prove beneficial in certain patient populations, for whom they may be the only option, but additional studies are required.
Myokines and monoclonal antibodies
One of most intensively studied therapeutic targets of muscle atrophy is myostatin, a well-known myokine having negative effects on muscle regeneration and mass [130]. Although many candidates (mostly monoclonal antibodies or soluble receptors) blocking myostatin-activin receptor signaling pathway have failed or been withdrawn from clinical trials, it is still one of the most promising and pursued pipelines. For instance, bimagrumab (BYM338), a human monoclonal antibody antagonizing the activin receptor type 2B, was developed by MorphoSys AG (Munich, Germany) and Novartis (Basel, Switzerland) and improved lean body mass, muscle volume, and 6-minute walk test results, but was withdrawn from phase 2 clinical trials for sporadic inclusion body myositis (sIBM) (Table 2, Fig. 2) [131]. Unfortunately, other monoclonal antibodies or peptides targeting myostatin-activin receptor signaling pathways, such as trevogrumab (REGN1033, Regeneron Pharmaceuticals Inc. and Sanofi SA), garetosmab (REGN2477, Regeneron Pharmaceuticals Inc. and Sanofi SA), ACE-083 (Acceleron), ACE-2494 (Acceleron), RG6206 (RO7239361, Roche and Genentech), domagrozumab (PF06252616, Pfizer), and sarconeos (BIO101, Biophytis) have not yet reached the primary endpoint (Table 2).
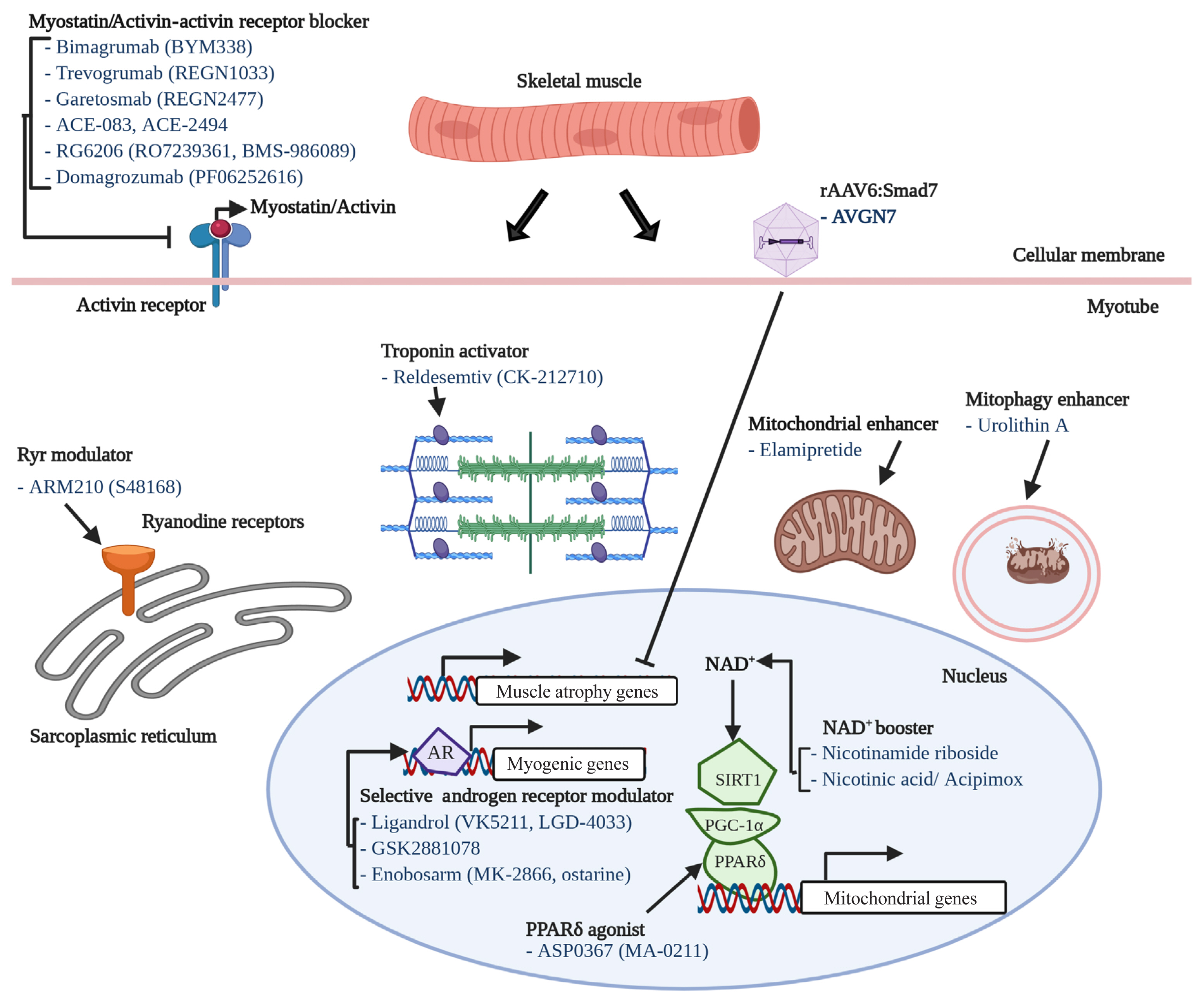
Potential factors underlying muscle aging. AR, anabolic resistance; NAD+, nicotinamide adenine dinucleotide; GDF, growth differentiation factor.
As aforementioned, recent studies have discovered several potential candidates associated with muscle atrophy or aging, such as GDF11 and GDF15. Recently, several biotechnology and pharmaceutical companies have adopted new strategies to develop novel therapeutics such as a dual agonist, a triple agonist, dual antagonists, and so forth [132,133]. Those kinds of concepts must also be explored in the field of muscle aging.
NAD+ boosters
Herein, we define NAD+ boosters as any therapeutic preserving subcellular NAD+ content through increasing/maintaining the NAD+ de novo biosynthesis pathway and salvage cycles, or the inhibition of NAD+ consumption enzymes. NAM, NMN, NR, and nicotinic acid (NA, also known as niacin) are the most intensively studied NAD+ boosters and are integral to the NAD+ biosynthesis pathways [52]. We have reported that oral supplementation of NR restored the NAD+ pool, improved the muscle stem cell pool, and improved muscle function in aged mice and a murine model of Duchenne muscular dystrophy (DMD; i.e., mdx mice) [51,55]. Consistent with these findings, Elhassan et al. [53] reported that NR supplementation (1,000 mg/day) increased NAD+ in skeletal muscle of older adults without obesity (median 75 years old; body mass index <30 kg/m2) and decreased circulating levels of pro-inflammatory cytokines. In contrast, Dollerup et al. [134], administered 2,000 mg/day of NR to obese men and did not observe any alterations in NAD+ levels or mitochondrial gene expression in men. Mills et al. [135] demonstrated that 12-month administration of NMN increased skeletal muscle NAD+ levels, elevated physical activity, increased energy expenditure, improved mitochondrial respiration, decreased age-associated weight gain, improved eye function and increased bone mineral density. Guo et al. [136] found that NAM protected streptozotocin-induced diabetic mice from muscle atrophy. More than 50 trials of NAD+ boosters are listed on the U.S. National Library of Medicine Clinical Trials website (https://clinicaltrials.gov/). The outcomes of ongoing and planned human trials will be critical to confirming or refuting the therapeutic potential of NAD+ boosters for sarcopenia.
Sarcopenia pipeline
We have described several potential therapeutics for sarcopenia from recent publications. Nineteen drug candidates in development for treating sarcopenia are summarized in Table 2. Their modes-of-action are also included in Table 2, Fig. 2. Eight of the 19 drug candidates are monoclonal antibodies or peptides targeting myostatin or the activin receptor, eight are small molecules targeting the androgen receptor, ryanodine receptors (RyRs), fast skeletal muscle troponin complex (TnT/TnI/TnC), PPARδ, or mitophagy activation, and one is a peptide targeting mitochondria function.
CONCLUSIONS
Nowadays, in the context of increasing average life expectancy, sarcopenia and frailty deserve more attention from researchers. Although many international and regional organizations have provided guidelines for assessing sarcopenia, countries still need to develop their own diagnostic toolkits following the anthropometric characteristics and medical capabilities of each country.
Although many recent studies have expanded our understanding of sarcopenia, these findings were primarily from animal models or correlative biomarkers in the human blood and feces, indicating the limitation of our knowledge on sarcopenia. Performing randomized control trials in humans to test these therapeutics and modalities are critical and the lack thereof could be a reason for the inconsistent results between studies. However, the difficulty in obtaining muscle biopsies from those suffering from muscle atrophy may mean we must develop non-invasive or minimally invasive methods. For instance, fluxomics which can measure markers of muscle protein synthetic rate from saliva, urine, or blood of human patients and magnetic resonance spectroscopy, which monitors mitochondrial oxidative capacity, may be suitable methods for patients suffering from sarcopenia [137–140]. In addition, longitudinal monitoring studies spanning age, gender, and race have to be done to find early predictive biomarkers for the risk of sarcopenia.
In addition to non-invasive human studies, animal model-based studies also have to be performed using modern multi-omics technology combined with computational analyses to address underlying cellular and molecular mechanisms, categorize sarcopenia into subclass based on revealed hidden signatures, validate biomarkers, and apply precision medicine for the prevention and treatment of sarcopenia.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2020R1A2C2010964) to Dongryeol Ryu and grants from the Canadian Institutes of Health Research (MOP 159455) and the Natural Sciences and Engineering Research Council of Canada (RGPIN 2018-06838, DGECR 2018-00012) to Keir J. Menzies. Alexander E. Green is the recipient of a uOttawa Eric Poulin Centre for Neuromuscular Disease (CNMD) Scholarship in Translational Research (STaR) Award which is supported by the University of Ottawa Brain and Mind Research Institute (uOBMRI).