Recent Improvements in Genomic and Transcriptomic Understanding of Anaplastic and Poorly Differentiated Thyroid Cancers
Article information

Abstract
Anaplastic thyroid cancer (ATC) is a lethal human cancer with a 5-year survival rate of less than 10%. Recently, its genomic and transcriptomic characteristics have been extensively elucidated over 5 years owing to advance in high throughput sequencing. These efforts have extended molecular understandings into the progression mechanisms and therapeutic vulnerabilities of aggressive thyroid cancers. In this review, we provide an overview of genomic and transcriptomic alterations in ATC and poorly-differentiated thyroid cancer, which are distinguished from differentiated thyroid cancers. Clinically relevant genomic alterations and deregulated signaling pathways will be able to shed light on more effective prevention and stratified therapeutic interventions for affected patients.
INTRODUCTION
Thyroid cancer is one of the most common malignancy in human endocrine systems [1]. The most prevalent types of thyroid cancer are differentiated thyroid cancers (DTCs) which are developed from follicular cells of thyroid [2]. Papillary thyroid cancer (PTC; 80% to 85%) and follicular thyroid cancer (FTC; 10% to 15%) account for the majority of thyroid cancers and most patients have good prognosis [23]. However, patients with advanced forms of thyroid cancer including poorly-differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) show worse prognosis compared with DTC [4]. Among them, ATC is the most fatal human cancer type with a median survival of 5 months but there is no effective therapeutic option for this fatal disease yet [4].
Through the recent surge of next-generation sequencing (NGS) technology, a myriad number of studies have been successfully deciphered the underlying molecular nature of human cancers [56]. Since various genetic factors affect therapeutic vulnerability or resistance to drugs [789], understanding molecular events occurred during thyroid cancer progression would provide effective therapeutic strategy. Also, the early detection of those markers in DTC is important to prevent and predict the progression. In this review, we focus on key genomic and transcriptomic events in aggressive thyroid cancers discovered in NGS era.
GENOMIC HALLMARKS
BRAF and RAS family genes
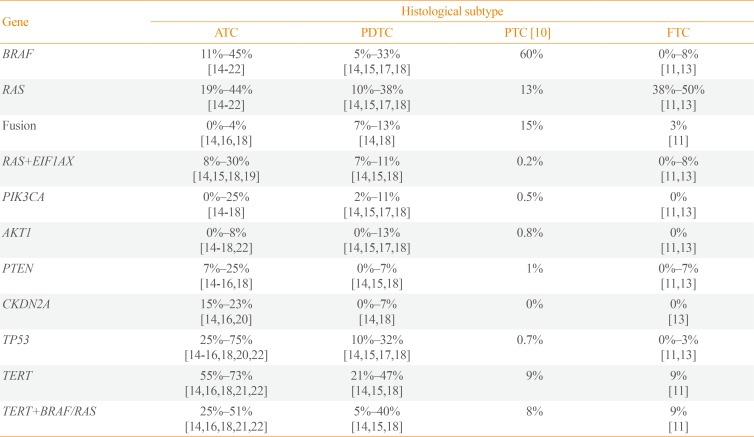
BRAF is a proto-oncogene encoding a serine/threonine-protein kinase and altered in 69% to 71% of PTC (classical type) [1011]. This gene has a crucial role in the Ras/Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway and its alterations affect cell cycle and progression [12]. The Ras oncogene family (hereafter referred as RAS) including HRAS, KRAS, and NRAS is most commonly altered in follicular-patterned thyroid tumors (follicular adenoma [FA], FTC, and follicular variant of PTC) [101113]. Since BRAF and RAS are tightly associated with the histology of thyroid cancer, they could be identifiers for tumor origin of ATC.
In ATC, BRAF and RAS mutations were identified with frequencies of 11%–45% and 19%%44%, respectively (Table 1) [141516171819202122]. Those alterations usually displayed similar incidence or sometimes BRAF was slightly more frequent than RAS [141516171819]. There is one study describing exceptionally high frequency of BRAF (91%) [23], and a few studies reported that RAS was more common than BRAF [172122]. In general, the combined frequency of BRAF and RAS mutations accounts for more than 50% of ATCs which signifies that ATC is commonly originated from DTC rather than de novo [14151618192223]. It is also reported that 5%–33% and 10%–38% of PDTCs harbor BRAF and RAS mutations, respectively [1418].

Frequency of Commonly Altered Genes in Aggressive Thyroid Cancers
Fusion gene rearrangement
It has been reported that ATC is barely developed with fusion gene. According to Landa et al. [18], only one out of 33 ATCs had a fusion gene, but 13% of PDTC had well-known thyroid cancer driver fusions (five RET, three paired box 8 [PAX8]-peroxisome proliferator activated receptor gamma [PPARG], and three ALK receptor tyrosine kinase [ALK] fusions). In PDTC, ALK rearrangement is known to be relatively common; 16% of thyroid tumors with ALK rearrangement are PDTC [2425]. Intriguingly, striatin (STRN)-ALK is predominantly found in thyroid cancer including PDTC rather than EMAP like 4 (EML4)-ALK, the most well-known ALK rearrangement. A recent in vivo study showed that PDTC is frequently developed in 22% and 36% of thyroglobulin (Tg)-STRN-ALK mice with and without goitrogen treatment, respectively [26]. In a study with the largest cohort of 196 ATCs, only 4% of ATCs harbored fusion genes including three BRAF (two with KIAA1549 and one with epidermal growth factor receptor pathway substrate 15 like 1 [EPS15L1]), two neurotrophic receptor tyrosine kinase 1 (NTRK1) (with lamin A/C [LMNA] and tropomyosin 3 [TPM3]), and three coiled-coil domain containing 6 (CCDC6)-RET fusions [16]. The low prevalence of fusion gene in ATC is also confirmed by genomic profiling of thyroid cancer cell lines. There were only two out of 31 ATC cell lines with oncogenic fusions including makorin ring finger protein 1 (MKRN1)-BRAF (in THJ1-6T) and fibroblast growth factor receptor 2 (FGFR2)-oxoglutarate dehydrogenase (OGDH) (in THJ-29T) [27].
EIF1AX
Eukaryotic translation initiation factor 1A X-linked (EIF1AX) encodes an essential eukaryotic translation initiation factor and is recently proposed as a cancer driver gene. Its role in driving cancer is firstly reported in uveal melanoma [28]. EIF1AX is usually co-mutated with G protein subunit alpha q (GNAQ) or G protein subunit alpha 11 (GNA11), and melanoma patients with EIF1AX mutations have good prognosis compared with others [29]. From pan-cancer data, only 0.3% of tumors (33/10,967) harbor hotspot mutations in EIF1AX (from http://www.cbioportal.org) [30]. Until now, its role in human cancer is not fully investigated, but in vitro analysis showed that increased EIF1AX activity triggers protein translation and cell proliferation [31]. In The Cancer Genome Atlas (TCGA) study, EIF1AX was confirmed as a driver gene of PTC (mostly for follicular variant types) [10]. It is also altered in FA and minimally invasive FTC [1113], which signifies less aggressive nature of EIF1AX. In contrast to uveal melanoma, EIF1AX mutation is sole event and does not cooperate other mutation in DTC. It is mutually exclusive with other driver mutations such as BRAF, RAS, and fusion genes [101113].
However, the mutually exclusiveness of EIF1AX to other driver genes is weaken and it is often co-occurred with RAS in aggressive thyroid cancers. According to Kunstman et al. [19], all of tumors with EIF1AX also harbored RAS mutations (NRAS or KRAS), and it accounts for 50% of RAS-positive ATCs. Furthermore, several studies with the larger cohorts confirmed the prevalence of EIF1AX+RAS in PTC, widely invasive FTC (wiFTC), PDTC, and ATC as 0.2%, 17%, 7%–11%, and 8%–30%, respectively [10141518]. In particular, EIF1AX-A113splice variant was commonly identified in EIF1AX-RAS co-mutated tumors [141832]. A recent experimental analysis suggested that protein synthesis in EIF1AX-A113splice knock-in thyroid cancer cell lines is increased through an activating transcription factor 4 (ATF4)-induced dephosphorylation of eukaryotic initiation factor 2 alpha (EIF2α) [32]. Furthermore, EIF1AX promotes the mammalian target of rapamycin (mTOR) activation to amino acid supply through cooperation of ATF4 and cellular myelocytomatosis oncogene (c-MYC). This study also showed that combinational treatment of mTOR kinase inhibitor (AZD8055) with either MEK inhibitor (trametinib) or bromodomain-containing protein 4 (BRD4) inhibitor (JQ1) to EIF1AX-A113splice knock-in CAL62 cell line resulted in huge tumor reduction and decreased c-MYC and mTOR protein levels.
Meanwhile, cyclin E1 (CCNE1) amplification rather than EIF1AX mutation was also reported to be occurred with 4% of RAS-positive ATCs [1620]. In those reports, targeted sequencing platform from Foundation Medicine Inc. was applied which does not include EIF1AX for genomic profiling. Therefore, the implication of CCNE1 and its relationship to EIF1AX or RAS is needed to be further investigated.
PIK3CA, AKT1, and PTEN
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and AKT serine/threonine kinase 1 (AKT1) are members of the oncogenic phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling pathway which are altered in diverse human malignancies [33]. The prevalence of PIK3CA mutations in human cancer is varied across cancer types: e.g., endometrial carcinoma (37%), breast cancer (31%), colorectal carcinoma (17%), pancreatic carcinoma (3%), and melanoma (2%) [34]. On the other hand, AKT1 is less frequently altered in human cancers relative to PIK3CA, and most commonly mutated in adenoid cystic carcinoma (4%), endometrial carcinoma (3%), and breast cancer (3%) [34]. In PTC, the prevalence of PIK3CA and AKT1 mutations are known to be 0.5% and 0.8%, respectively [10], and they are not reported in follicular thyroid tumors [1113]. Moreover, they are slightly frequent in metastatic PTC (3% for both) compared with PTC [14]. However, several reports showed that PIK3CA and AKT1 are frequently altered in aggressive thyroid cancers. The frequency of PIK3CA mutation in PDTC and ATC is 2%–11% and 0%–25%, respectively [1415161718], and AKT1 (especially for E17K) is found in 0%–13% and 0%–8% of PDTC and ATC, respectively [1415161718]. It has been reported that PIK3CA mutations are frequently co-occurred with BRAF or RAS in diverse types of advanced cancers [353637]. However, PIK3CA mutations are usually reported as co-existed with BRAF in aggressive thyroid cancers [1415161718192338], and only a few studies showed that RAS-PIK3CA co-mutation is more frequent than BRAF-PIK3CA co-mutation [2122]. In agreement with other cancer types, AKT1E17K is less common compared with PIK3CA, but it is also co-occurred with BRAF [141622]. Moreover, PIK3CA and AKT1 mutations are completely mutually exclusive to each other as reported in other cancer types [39]. It suggests that those mutations may have biologically equivalent role in activating PI3K/AKT/mTOR pathway to drive progression of thyroid cancer [4041].
Furthermore, high prevalence of phosphatase and tensin homolog (PTEN) alteration (7% to 25%) which regulates PI3K/AKT/mTOR signaling pathway was also reported in aggressive thyroid cancers [1415161820222342]. Notably, two studies reported that most of PTEN alterations (45%, 10/22) was co-occurred with neurofibromin 1 (NF1) or RB transcriptional corepressor 1 (RB1) rather than BRAF or RAS [1618].
TERT
Telomerase activation is one of the hallmarks of cancer [43]. The activation of telomerase reverse transcriptase (TERT) is usually induced by the mutations in the promoter region known as C228T or C250T (96% in pan-cancer) [44]. In DTC, several reports confirmed its significance in poor survival of the patients [454647]. Especially, when TERT promoter mutations co-exist with BRAFV600E or RAS mutations, they exert a negative synergistic effect on prognosis. Several studies demonstrated the potential mechanism of this synergism between the mutations; BRAFV600E-induced E26 transformation-specific or E-twenty-six (ETS) transcription factors increase TERT expression by binding to the ETS-binding site generated by TERT promoter mutation [4849]. The frequency of TERT promoter mutations in PTC are reported as 9% [10], but their incidences are greatly increased in metastatic PTC (60%) [44]. Moreover, 21% to 47% PDTC and 55% to 73% ATC are known to have TERT promoter mutation that again emphasize the critical role of TERT in thyroid cancer progression [141516182122]. Recently Yoo et al. [14] reported the novel types of TERT alterations in wiFTC including fusion gene and translocation in TERT upstream, and they also induced extremely high expression levels of TERT. The rearrangements with/within TERT are often identified in aggressive human cancer types [505152]. Therefore, TERT rearrangements in thyroid cancer are also significant events that can induce the progression. Moreover, previously analyzed aggressive thyroid cancers without TERT promoter mutations more likely to harbor unidentified TERT rearrangements since these events are mutually exclusive to TERT promoter mutations and most studies did not focus on intergenic region [1453].
TP53
Tumor protein p53 (TP53) is the most well-studied human tumor suppressor and its incidence is reported as 37% in pan-cancer data (3,786/10,225) [54]. More than 90% of patients in some cancer types, such as ovarian cancer and uterine carcinosarcoma, harbor mutation in TP53, but the seven cancer types have incidence less than 5% (0.7% for DTC) [1011]. Mutations in this gene is tightly associated with the progression and poor prognosis of human cancers including ATC [55565758]. In contrast to DTC, its prevalence is extremely increased according to the aggressiveness of thyroid cancer; the frequency of TP53 in PDTC and ATC were reported as 10%–32% and 25%–75%, respectively [14151617182122]. Since TP53 alterations are relatively rare in metastatic PTC (13%) and wiFTC (8%) which are early advanced forms of DTC whereas TERT promoters are commonly mutated in them (48% and 75% for each), it is suspected that TERT and TP53 are major players triggering early and late progression of thyroid cancer, respectively (Fig. 1) [14].
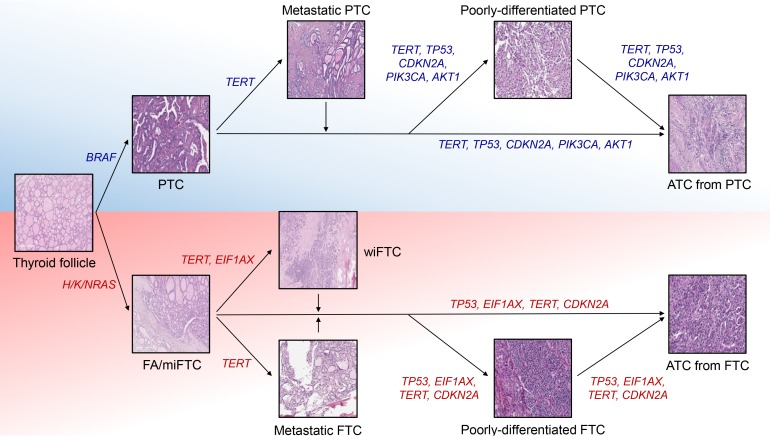
The major genetic contributors to thyroid cancer progression. Progression mechanisms of BRAF-positive papillary thyroid cancer (PTC) and RAS-positive follicular thyroid cancer (FTC) are illustrated. TERT, telomerase reverse transcriptase; TP53, tumor protein p53; CDKN2A, cyclin dependent kinase inhibitor 2A; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; AKT1, AKT serine/threonine kinase 1; ATC, anaplastic thyroid cancer; EIF1AX, eukaryotic translation initiation factor 1A X-linked; FA, follicular adenoma; miFTC, minimally invasive FTC; wiFTC, widely invasive FTC.
CDKN2A
Cyclin dependent kinase inhibitor 2A (CDKN2A) encodes a tumor suppressor protein, p16INK4a, which has a critical role in the inhibition of cell cycle progression [59]. This gene is located at 9q21.3 locus and is known to be deactivated by diverse mechanisms such as homozygous loss, exon skipping, truncating mutations, and epigenetic silencing [60]. In particular, loss of CDKN2A and its significance in cancer progression have been well reported in several types of cancers [616263]. However, its association to thyroid cancer progression is recently proposed by a few studies. There are three reports that discovered CDKN2A loss in aggressive thyroid cancers, and its incidence in ATC was reported as 15% to 23% [141620]. In addition, CDKN2A deletion is also frequent in thyroid cancer cell lines (47%) [27]. It could be robustly associated with thyroid cancer progression since this type of variation is not identified in DTC [1013]. According to Yoo et al. [14], CDKN2A loss is not only associated with the progression of ATC, but also involved in poor prognosis and survival of patient with advanced DTC and ATC. By thyroid differentiated score (TDS) analysis from TCGA [10], the poorer differentiation status in ATCs with CDKN2A loss relative to those with CDKN2A2n was identified. Furthermore, patients with ATC or advanced DTCs harboring CDKN2A/p16 loss showed poor disease-specific survival. Hence, CDKN2A can be used as a useful biomarker to monitor the prognosis of thyroid cancer patients.
Other genomic characteristics
In addition to mutation profiles, some reports described the additional genomic features related to aggressive thyroid cancers. A recent study reported that one out of eleven ATCs showed tendency to microsatellite instability high (MSI-H) [64]. There is a report that also showed MSI in the synchronous FTC (MSI borderline), PDTC, and ATC from one patient [65]. Moreover, some PDTC (T243 with MutS homolog 2 [MSH2] mutation) and FTC (FTC-133, FTC-236, and FTC-238 with MSH6, MSH6/MLH1, and MSH6 mutations, respectively) cell lines have mutations in mismatch repair genes and exceptionally high tumor mutational burden (TMB) [27]. Since patients with MSI-H, mismatch repair deficiency (MMR-D), or high TMB tumors show the durable clinical benefit from immunotherapy [66676869], this therapy could be a new option for the minor portion of patients with aggressive thyroid cancer. Also, the mutational signature analysis from two studies showed increased Apolipoprotein B mRNA Editing Catalytic Polypeptide-like (APOBEC) cytidine deaminase activity in aggressive thyroid cancers [1416]. As an APOBEC-related mutagenesis also leads to high TMB and linked to the immune activations [7071], patients with this mutational signature would be also potential target of immunotherapy.
TRANSCRIPTOMIC HALLMARKS
Molecular subtype
Until now, only a small number of studies investigated transcriptomic nature of aggressive thyroid cancers. Landa et al. [18] showed the transcriptomic feature of ATC using TCGA's BRAFV600E-RAS score (BRS) analysis [10]. From the original investigation of TCGA, DTC could be sub-classified into BRAFV600E-like (with negative BRS) and RAS-like (with positive BRS) (Fig. 2A). In general, BRAFV600E-like and RAS-like are associated with BRAF and RAS mutations, respectively, and BRAFV600E-like DTCs display more aggressive clinical characteristics than RAS-like DTCs [11]. However, ATC did not follow the BRS rules, but most of them showed BRAFV600E-like signature (negative BRS) although some of them harbored RAS mutations [18]. Yoo et al. [14] also reported a consistent result using BRS analysis (negative BRS for both BRAF and RAS mutated ATCs) and also proposed advanced view of transcriptomic feature. Based on principal component analysis, BRAF-positive and RAS-positive ATCs showed similar expression profile as in BRS analysis, but they were grouped into novel molecular subtype, ATC-like, rather than BRAFV600E-like. This result signifies that ATCs share similar global transcriptomic features regardless of their mutational status and have totally distinctive molecular characteristics from DTC.

Transcriptomic signatures of thyroid cancer. (A) Transcriptome based molecular subtype classifications of thyroid cancer according to histological subtypes. From The Cancer Genome Atlas (TCGA)'s original investigation, papillary thyroid cancers are classified into two molecular subtypes, BRAFV600E-like and RAS-like [10]. Afterward, Yoo et al. [11] showed that RAS-like can be breakdown into RAS-like and non-BRAF/non-RAS subtype (NBNR). RAS-like tumors with eukaryotic translation initiation factor 1A X-linked (EIF1AX), paired box 8 (PAX8)-peroxisome proliferator activated receptor gamma (PPARG), and THADA armadillo repeat containing (THADA) fusion were re-classified into NBNR. Dicer 1, ribonuclease III (DICER1), enhancer of zeste 1 polycomb repressive complex 2 subunit (EZH1), isocitrate dehydrogenase (NADP(+)) 1 (IDH1), and speckle type BTB/POZ protein (SPOP) are also associated with NBNR signature. (B) Schematic illustration of activated and deactivated signaling pathways according to the aggressiveness of thyroid cancer. BRS, BRAFV600E-RAS score; MAPK, mitogen-activated protein kinase; ECM, extracellular matrix; PD, programmed death; VEGF, vascular endothelial growth factor; JAK-STAT, Janus kinase-signal transducer and activator of transcription.
Intracellular signaling pathways
There are two studies conducted on differentially expressed genes (DEGs) and pathways analyses in ATC (Fig. 2B). Kasaian et al. [72] identified that extracellular matrix (ECM)-receptor interaction, focal adhesion, cell cycle, p53, and general cancer signaling pathways were up-regulated in ATC correspondence to PTC and normal thyroid tissue [72]. Moreover, cancer-related genes such as MYC, MTOR, protein kinase C alpha (PRKCA), and transforming growth factor beta 1 (TGFB1) were overexpressed. Meanwhile, tight junctions, cell adhesion molecules, various metabolism pathways, and thyroid differentiation signature genes such as TG, transcription termination factor 1 (TTF1), thyroid stimulating hormone receptor (TSHR), and thyroid peroxidase (TPO) were decreased. Yoo et al. [14] also discovered similar transcriptome changes in ATC. DEG and pathway analyses were performed separately using ATC and DTC based on driver mutation status (BRAF and RAS). This study showed that both BRAF-positive and RAS-positive ATCs displayed up-regulation of the mitogen-activated protein kinase (MAPK), ECM-receptor, focal adhesion, cell cycle, p53, and general cancer signaling pathways as Kasaian et al. [72] reported. Also, BRAF-positive and RAS-positive ATCs showed activation of vascular endothelial growth factor (VEGF)/notch and Janus kinase (JAK)-Signal transducers and activators of transcription (STAT) signaling pathways, respectively. In particular, suppressor of cytokine signaling 3 (SOCS3), BCL2 like 1 (BCL2L1), and MYC were up-regulated in RAS-positive ATCs, and in vitro experiments showed that their expression level and cell proliferation were decreased upon treatment of ruxolitinib, JAK inhibitor. Furthermore, several metabolism pathways and most of thyroid differentiation genes included in TDS analysis were down-regulated in ATC.
Immune signature
Understanding immune cell signature in human cancer is one of the important field in cancer biology since they are closely involved in the cancer progression or therapeutic response [73]. Also, the high degree of immune cell infiltration is reported to be associated with unfavorable clinical outcomes of patients with DTC [74]. Landa et al. [18] showed that ATC have elevated M2 macrophage infiltration compared with PDTC based on the expression profile of 68 genes which are subset of 78 genes associated with M2 macrophages [1875]. Giannini et al. [76] also displayed increased macrophage score in ATC compared with PDTC, PTC, and normal thyroid. Moreover, tumor infiltrating leukocyte, T-cells (CD8+ and exhausted CD8+ T-cells), and cytotoxicity cell scores were significantly elevated in ATC. Interestingly, aforementioned immune cell infiltrations were less active in PDTC relative to PTC. In addition, two types of immune signatures, ATC-like and PDTC-like, in thyroid cancer were found. They investigated four categories of immune signatures: hot, altered-immunosuppressed, altered-excluded, and cold [77]. According to the study, most of ATCs were defined as two main immune contextures, hot (34%; T-cell-inflamed) and altered-immunosuppressed (50%; a low degree of T-cell infiltration, but presence of soluble inhibitory mediators, immune suppressive cells, and T-cell checkpoints). On the other hand, PDTC showed another two immune contextures: cold (65%; non-T-cell-inflamed) and altered-excluded (14%; T-cell at the invasive margins or tumor edge without intratumoral infiltration).
In addition to immune cell infiltration, the up-regulation or amplification of CD274 (encodes programmed cell death 1 ligand 1 [PD-L1]) or PDCD1LG2 (encodes programmed cell death 1 ligand 2 [PD-L2]) which belong to the family of immune checkpoint proteins were reported in a handful of ATCs [1416]. There have been also various reports showed expression of PD-1 or PD-L1 in ATC using immunohistochemistry [78798081]. These markers are considered as imperfect predictor of immunotherapy response alone [82], but combination of their expression and other genomic features (MSI-H, MMR-D, or TMB) could be better predictor for immunotherapy response of patients with ATC.
CONCLUSIONS
Herein, we reviewed the recent findings in NGS era regarding the genomic and transcriptomic changes associated with ATC and PDTC. The key molecular features of ATC could be shortened as follows. First, co-mutations of TP53 (25% to 75%) as well as TERT (55% to 73%) are hallmarks of ATC considering their remarkably high prevalence. Second, the additional mutational hits in oncogenes such as PIK3CA/AKT1 (with BRAF) or EIF1AX (with RAS). Third, BRAF-positive and RAS-positive ATCs display similar global transcriptome features, ATC-like. Fourth, the extra activations of cancer related signaling pathways such as ECM-receptor interaction, focal adhesion, cell cycle, p53, and MAPK. At last, dysfunction of thyroid hormone and various metabolism signaling pathways.
Despite the extensive molecular profiling of thyroid cancers, only limited number of studies regarding PDTC was reported yet. In particular, none of literature performed comprehensive DEG and pathway analyses including PTDC as well as ATC and DTC. Based on the findings from Giannini et al. [76], there might be dynamic molecular changes rather than linear progression as histological grades from DTC through PDTC to ATC. Hence, investigating this molecular feature would allow us to deeper understanding of molecular pathogenesis of thyroid cancer and improve patient care.
ACKNOWLEDGMENTS
This study was funded by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Science, ICT & Future Planning (NRF-2019R1A2C2084332).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.