
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 35(1); 2020 > Article
-
Brief ReportNovel ABCD1 Gene Mutation in a Korean Patient with X-Linked Adrenoleukodystrophy Presenting with Addison's Disease
-
Yun Kyung Cho1,2
 , Seo-Young Lee3, Sang-Wook Kim1
, Seo-Young Lee3, Sang-Wook Kim1 -
Endocrinology and Metabolism 2020;35(1):188-191.
DOI: https://doi.org/10.3803/EnM.2020.35.1.188
Published online: March 19, 2020
1Department of Internal Medicine, Kangwon National University School of Medicine, Chuncheon, Korea.
2Department of Internal Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
3Department of Neurology, Kangwon National University School of Medicine, Chuncheon, Korea.
- Corresponding author: Sang-Wook Kim. Department of Internal Medicine, Kangwon National University School of Medicine, 156 Baengnyeong-ro, Chuncheon 24289, Korea. Tel: +82-33-258-9169, Fax: +82-33-258-2455, sangwookkim@kangwon.ac.kr
Copyright © 2020 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- X-linked adrenoleukodystrophy (X-ALD) occurs due to mutations in the ABCD1 gene that encodes the peroxisomal membrane protein peroxisomal transporter ATP-binding cassette sub-family D member 1 (ABCD1). Degradation of very long-chain fatty acids in peroxisomes is impaired owing to ABCD dysfunction, subsequently leading to adrenomyeloneuropathy, cerebral adrenoleukodystrophy, and adrenal insufficiency. X-ALD frequently induces idiopathic Addison's disease in young male patients. Here, we confirmed the diagnosis of X-ALD in a young male patient with primary adrenal insufficiency, and identified a novel ABCD1 gene mutation (p.Trp664*, c.1991 G>A).
- X-linked adrenoleukodystrophy (X-ALD; OMIM:300100) is the most common peroxisomal disorder arising from mutations in the ABCD1 gene (located on Xq28) that codes for ATP-binding cassette sub-family D member 1 (ABCD1), the peroxisomal transporter for very long chain fatty acids (VLCFAs) [1]. Impairment in ABCD1 causes peroxisomal beta-oxidation deficiency of VLCFAs, and the deposition of VLCFAs in plasma and tissues [1]. Accumulation of VLCFAs contributes to the demyelinating pathology in adrenomyeloneuropathy (AMN), and cerebral neuropathy in X-ALD [1]. X-ALD also affects the adrenal glands. Primary adrenocortical insufficiency occurs in at least 70% of patients with X-ALD, and glucocorticoids are more likely to be affected compared to mineralocorticoids [1]. Young male patients with X-ALD can have adrenal insufficiency as the first, and often the only symptom [2]. Here, we confirmed the diagnosis of X-ALD in a young male patient with primary adrenal insufficiency (PAI), and identified a novel ABCD1 gene mutation (p.Trp664*) using whole-exome sequencing.
INTRODUCTION
- Clinical presentation
- In April 2014, a 20-year-old patient (the proband) was referred to the endocrinology and metabolism clinic with the diagnosis of idiopathic adrenal insufficiency (Addison's disease). Four months ago, he was diagnosed with adrenal crisis with community-acquired pneumonia while in military service. His face showed entire darkening of skin, and pigmentation on the vermilion border of lips. He also had hyperpigmented macules on palms, and slight darkening of the palmar creases (Fig. 1A). In the morning, plasma adrenocorticotropic hormone (ACTH) was 550.4 pg/mL (reference range, 10 to 60), and serum cortisol level was 4.4 µg/dL. Rapid ACTH stimulation test showed serum cortisol level to be 5.5 µg/dL at 60 minutes after stimulation. Plasma renin activity was 0.24 ng/mL/hr and serum aldosterone level was less than 1.0 ng/dL. Anti-adrenal cortex antibody was negative. Abdominal computed tomography scan did not show evidence of adrenal calcification or hemorrhage. He reported that he had general weakness since 5 years, and had noted the darkening of skin from the time of elementary school (3rd grade). Upon neurological examination, bilateral patellar and ankle reflexes were not abnormal. There was no Babinski's sign or ankle clonus. In magnetic resonance imaging (MRI) scans (T1-weighted image [T1WI], T2WI, and fluid attenuated inversion recovery), there was no significant lesion (Fig. 1B). VLCFA level analysis, showed increased levels of C26, C26/C22, and C24/C22 ratio in the serum (Table 1). His mother, elder brother, and elder sister denied any neurological symptoms or disabilities, and unfortunately, refused to participate in the VLCFA analysis. The patient has been doing well with fludrocortisone (0.05 mg once a day) and hydrocortisone (20 to 10 mg twice a day), as of September 23, 2019 without any sign of AMN. Proband's mother is healthy, with no evidence of neurological disability.
- Ethics statement
- Written informed consent was obtained from the proband and his mother before analyzing the whole gene. Institutional Review Board (IRB) approval was not required since genetic analysis is an essential and routine diagnostic care of patients with X-ALD. Failure to do so would lead to inadequate diagnosis, making genetic counseling impossible. The current investigation was conducted under the adoption of the American College of Medical Genetics and Genomics (ACMG) policy statements [3].
METHODS
- Genetic analysis
- Targeted resequencing is more sensitive and accurate than whole exome sequencing (WES) for detecting mutations causing X-ALD; however, we could not use this method owing to the high cost and unavailability of service vendors in South Korea. Instead, we used an alternative strategy of implementing WES and subsequently confirming the results with Sanger sequencing. In December 2017, WES was performed with patient genomic DNA using the SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed using the HiSeq 2000 system (Illumina, San Diego, CA, USA) at Macrogen (Seoul, Korea).
- Whole-exome sequencing of both proband and his mother revealed the presence of a mutation generating stop codon at the position c.1991G>A (NM_000033.3, p.Trp664*). Using Sanger sequencing, the mutation was confirmed (Fig. 1C). This nonsense mutation had not been reported previously (The ALD Mutation Database, last modified on 2018-06-11, https://adrenoleukodystrophy.info/mutations-and-variants-in-abcd1). Based on the ALD Mutation Database, p.Trp664*, which also resulted from c.1992G>A, had been identified in a patient with ALD [4], and was reported to be deleterious. Our case was not showing AMN phenotype clinically, but was confirmed with Addison's disease. Biochemically, we also confirmed the increase of VLCFA. The identified genetic mutation was a stop codon that generated a truncated ABCD1 protein. An X-ALD case with the same stop codon-causing mutation has been registered in bioinformatics database. These constitute very strong evidence to suggest the identified variation as PVS1 (very strong evidence of pathogenicity) as outlined in the ACMG guidelines [5]. The variation also met PS1, PM2, PP3, and PP4 criteria according to the ACMG guideline [5]. Therefore, the variation that we discovered can be classified as pathogenic with 1 very strong and 1 strong, 1 moderate and 2 supporting evidences.
- The nonsense mutation was also predicted to result in a disease by affecting the encoded protein, as shown by MutationTaster (http://www.mutationtaster.org/).
RESULTS
- The incidence of adrenocortical insufficiency in X-ALD is reported to be about 70% [6]. Interestingly, in some cases, only adrenocortical insufficiency is present without neuropathy. Dubey et al. [7] had conducted a rapid ACTH stimulation test in a relative of a patient with X-ALD, who had no neurologic symptom. They found evidence of adrenocortical insufficiency even in absence of any neurological abnormality and in MRI. In the present case, color of the skin had darkened several years before the onset of adrenal crisis, and history of long-standing systemic weakness may be presumed to indicate the start of adrenocortical insufficiency at an early age.
- The reason behind reporting this case is that the mutation is novel, and not yet listed in any database such as ClinVar or X-ALD database. In the era of next generation sequencing, mutations that cause rare genetic diseases are becoming prevalent. By reporting the relationship between mutations of human genes and phenotypes, with supporting evidence, it will become feasible for clinicians to identify whether or not a mutation is pathological, likely, or completely unrelated to the disease. We believe that our report will eventually provide useful information to researchers studying X-ALD.
- Second, genetic counseling is an important aspect of X-ALD, which is inherited in an X-linked manner. Women with X-ALD very rarely develop adrenal insufficiency or cerebral involvement. Proband's mother, who was a carrier, negated any neurological symptom or disability, and clinical evidence of adrenal insufficiency. However, carrier females have a 50% chance of transmitting the ABCD1 pathogenic variant in each pregnancy. We have informed the patient's elder sister that she would need genetic counseling when pregnancy is planned, and she agreed to get tested for detection of mutation in future. Since genetic counseling can prevent the inheritance of a disease, clinicians should not restrict themselves in just diagnosing and treating genetic disorders, but should also find the causative variants and conduct genetic counseling within the family and relatives.
- Third, the prevalence of PAI in Korean population is significantly lower than that in Western countries [8]. The estimated incidence was 0.45 per million per year, and the etiology of PAI was not identified in 34.9% of cases. Adrenal insufficiency may be the initial presentation of X-ALD (Addison-only phenotype), and patients in this category might develop neurological symptoms later; therefore, it is important to consider X-ALD in any male patient presenting with Addison's disease.
- In conclusion, we report the case of a patient with X-ALD presenting with adrenal insufficiency, and have identified a novel ABCD1 gene mutation (p.Trp664*, c.1991G>A) using whole-exome sequencing.
DISCUSSION
-
Acknowledgements
- This study was supported by a Research Grant from the Korean Endocrine Society in 2016.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS:
Article information
- 1. Kemp S, Huffnagel IC, Linthorst GE, Wanders RJ, Engelen M. Adrenoleukodystrophy: neuroendocrine pathogenesis and redefinition of natural history. Nat Rev Endocrinol 2016;12:606–615. ArticlePubMedPDF
- 2. Huffnagel IC, Laheji FK, Aziz-Bose R, Tritos NA, Marino R, Linthorst GE, et al. The natural history of adrenal insufficiency in X-linked adrenoleukodystrophy: an international collaboration. J Clin Endocrinol Metab 2019;104:118–126. ArticlePubMedPDF
- 3. ACMG Board of Directors. Points to consider for informed consent for genome/exome sequencing. Genet Med 2013;15:748–749. ArticlePubMed
- 4. Salsano E, Tabano S, Sirchia SM, Colapietro P, Castellotti B, Gellera C, et al. Preferential expression of mutant ABCD1 allele is common in adrenoleukodystrophy female carriers but unrelated to clinical symptoms. Orphanet J Rare Dis 2012;7:10ArticlePubMedPMC
- 5. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. ArticlePubMedPMCPDF
- 6. Moser HW, Moser AB, Naidu S, Bergin A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci 1991;13:254–261. ArticlePubMed
- 7. Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr 2005;146:528–532. ArticlePubMed
- 8. Hong AR, Ryu OH, Kim SY, Kim SW. Korean Adrenal Gland and Endocrine Hypertension Study Group. Korean Endocrine Society. Characteristics of Korean patients with primary adrenal insufficiency: a registry-based nationwide survey in Korea. Endocrinol Metab (Seoul) 2017;32:466–474. ArticlePubMedPMC
References
(A) Hyperpigmented macules on palms, and slight darkening of the palmar creases. (B) Brain magnetic resonance imaging of the patient. No abnormal findings were noted in T2-weighted image (T2WI; left) and fluid attenuated inversion recovery (right) images. (C) Confirmation of hemizygous mutation of the patient, and heterozygous point mutation (p.Trp664*, c.1991G>A). Upper two rows of sequence chromatogram of the patient show the change of reference allele G to A, and lower two rows of his mother show the presence of both G and A alleles.
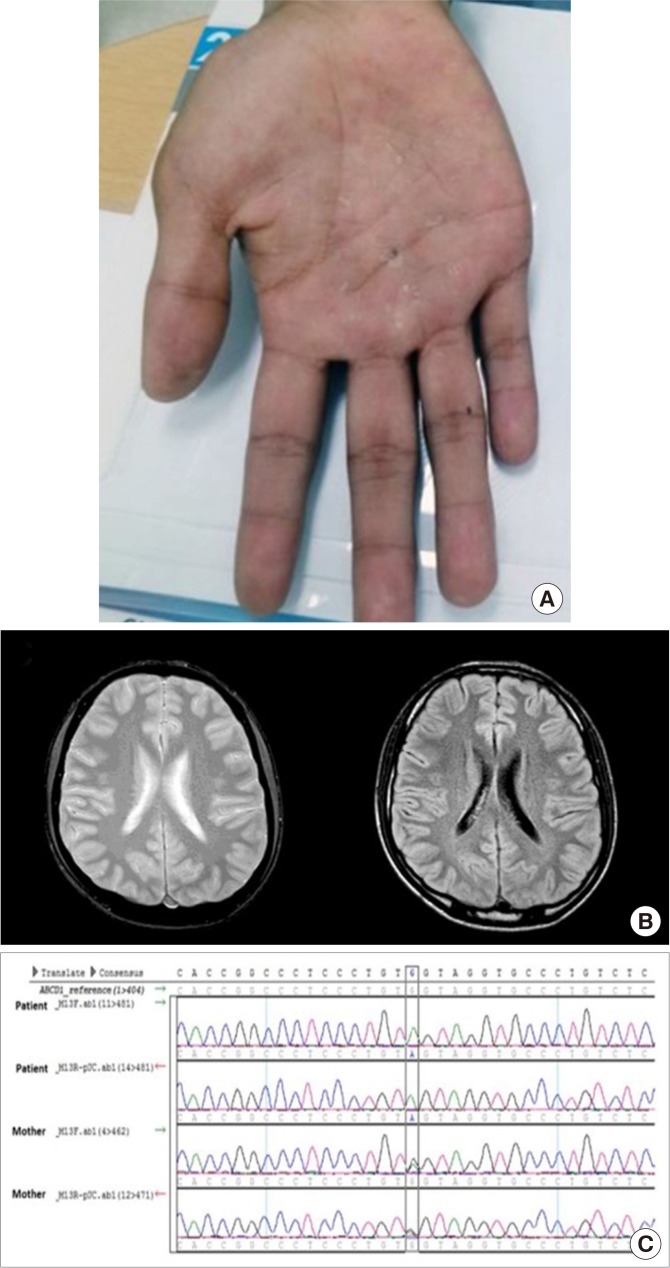
Figure & Data
References
Citations
- Ocular findings and genomics of X-linked recessive disorders: A review
Asima Hassan, YaserR Mir, RajaA H Kuchay
Indian Journal of Ophthalmology.2022; 70(7): 2386. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

