Radioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy
Article information

Abstract
The retained functionality of the sodium iodide symporter (NIS) expressed in differentiated thyroid cancer (DTC) cells allows the further utilization of post-surgical radioactive iodine (RAI) therapy, which is an effective treatment for reducing the risk of recurrence, and even the mortality, of DTC. Whereas, the dedifferentiation of DTC could influence the expression of functional NIS, thereby reducing the efficacy of RAI therapy in advanced DTC. Genetic alternations (such as BRAF and the rearranged during transfection [RET]/papillary thyroid cancer [PTC] rearrangement) have been widely reported to be prominently responsible for the onset, progression, and dedifferentiation of PTC, mainly through activating the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling cascades. These genetic alternations have been suggested to associate with the reduced expression of iodide-handling genes in thyroid cancer, especially the NIS gene, disabling iodine uptake and causing resistance to RAI therapy. Recently, novel and promising approaches aiming at various targets have been attempted to restore the expression of these iodine-metabolizing genes and enhance iodine uptake through in vitro studies and studies of RAI-refractory (RAIR)-DTC patients. In this review, we discuss the regulation of NIS, known mechanisms of dedifferentiation including the MAPK and PI3K pathways, and the current status of redifferentiation therapy for RAIR-DTC patients.
INTRODUCTION
Thyroid cancer has emerged as a striking health issue over recent decades because of its gradually increasing incidence worldwide. The global incidence of thyroid cancer is 6.7 per 100,000, and the number of newly diagnosed likely cases in China has exceeded 190,000 (194,232 cases) [1]. Papillary thyroid cancer (PTC), follicular thyroid cancer, and Hürthle cell cancer are derived from follicular cells; thus, they are collectively characterized as differentiated thyroid cancer (DTC), and account for more than 90% of all thyroid malignancies [23]. Although most DTC cases have a quite favorable prognosis after standard therapeutic approaches, including surgery, selective radioactive iodine (RAI) therapy, and thyroid stimulating hormone (TSH) suppressive therapy, the risk of local recurrence and distant metastasis may be up to 20% and 10%, respectively. Among these patients, two-thirds show initial or gradual loss of the ability of iodine uptake due to the dysfunction, and even loss, of sodium iodide symporter (NIS) expression in the basal membrane, indicating a status of dedifferentiation known as RAI-refractory DTC (RAIR-DTC), which is of major clinical concern because its 10-year survival rate is less than 10% [4].
Genetic alterations are the fundamental drivers for the tumorigenesis and pathogenesis of thyroid cancer, aberrantly activating the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways [56]. These alternations are also known to be associated with the silencing of various thyroid iodine-metabolizing genes, especially solute carrier family 5 member 5 (SLC5A5), which encodes NIS, thus resulting in the failure of RAI therapy [789]. Multiple agents have been investigated in attempts to restore NIS expression and to enhance RAI uptake in RAIR-DTC patients. This review aimed to summarize the regulation of NIS, the possible mechanism of NIS downregulation, and the potential of redifferentiation therapy for RAIR-DTC.
PHYSIOLOGICAL REGULATION OF NIS EXPRESSION
Iodide, as an essential component, is utilized by follicular cells to synthesize thyroid hormone in the normal thyroid. As it is expressed on the basal membrane, NIS provides the physiological basis for the active transport of iodide into the follicular cells of the thyroid [10]. DTC cells can retain similar functions to those of follicular cells, such as iodine uptake and iodination [1112], which allows RAI therapy to be the mainstay for the treatment of intermediate and high-risk DTC after surgery [13]. To destroy residual or potential subclinical lesions, RAI therapy could improve disease-specific survival and progression-free survival [14]. Hence, the function or expression of NIS in DTC cells is crucial for the efficacy of RAI therapy in such patients.
Functional NIS expression can be regulated at both the transcriptional and post-translational levels. As the predominant regulator of NIS expression, TSH is primarily involved at the translational level. After the binding of TSH with the TSH receptor, adenylyl cyclase is stimulated through the Gs-protein, which in turn increases the expression of cyclic adenosine monophosphate (cAMP). cAMP then induces NIS transcription by activating several signaling pathways that could stimulate the NIS upstream enhancer (NUE). It is now known that the human NUE consists of paired box gene-8 (PAX8; thyroid-specific transcription factor) binding site and cAMP-response element like site, both of which are important for the integrated activity of the NUE [15]. As illustrated in Fig. 1, cAMP can stimulate the NUE through both protein kinase A (PKA)-independent and -dependent pathways. Through redox effector factor-1 (Ref-1), the PKA-independent pathway, PAX8 is subsequently stimulated to bind to the NUE, leading to the activation of NUE [15161718], and this pathway plays a key role in the differentiation of the thyroid [19]. Through the PKA-dependent pathway, the activated PKA could phosphorylate the cAMP-responsive element modulator, enhancing NUE activity [2021].
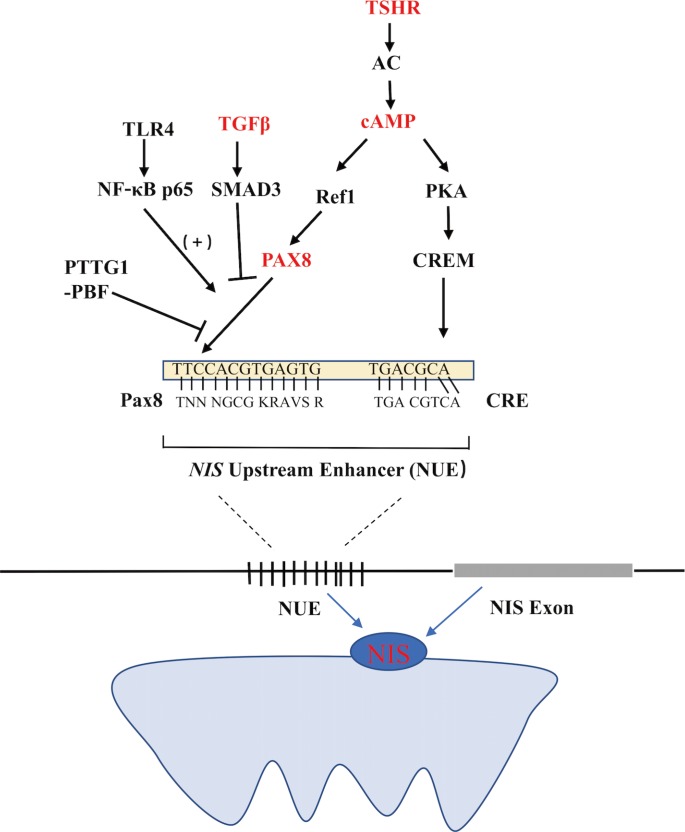
Regulation of the sodium iodide symporter (NIS) upstream enhancer (NUE) at the transcriptional level in thyroid cells. TSHR, thyroid stimulating hormone receptor; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CRE, cAMP-response element; CREM, CRE-modulator; Ref1, apurinic apyrimidinic endonuclease redox effector factor-1; Pax8, paired box gene-8; TGFβ, transforming growth factor β; TLR4, Toll-like receptor 4; NF-κB, p65, a member of the class II nuclear factor κ-light-chain-enhancer of activated B cells, p65; PTTG1, pituitary tumor-transforming gene-1; PBF, PTTG1-binding factor.
TSH-independent mechanisms also regulate NIS expression, which mainly include three pathways influencing the binding of PAX8 to NUE. First, in the transforming growth factor β (TGFβ)-SMAD signaling pathway, TGFβ activates the downstream Smad3 and subsequently inhibits the binding of PAX8 to NUE, significantly decreasing NIS mRNA expression in thyroid cells [916]. Second, in the Toll-like receptor (TLR)-nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, the TLR activates the downstream NF-κB, which further interacts with PAX8, activating NIS transcription via the NUE [2223]. Third, in the pituitary tumor-transforming gene-1 product (PTTG1)-binding factor (PBF) complex, the PTTG1 and the PBF could interfere with the binding of PAX8 to the NUE, thus suppressing the expression of NIS (Fig. 1) [242526]. Saez et al. [26] reported that increased PTTG1 expression could reduce the efficacy of RAI therapy in thyroid cancer.
Concerning post-translational regulation, abundant NIS expression may mis-localize in the intracellular compartment rather than the cell membrane [27]. This abnormal membrane targeting of NIS could disable iodide transport and result in the reduced uptake and accumulation of RAI in thyroid cancer cells, inducing the probable failure of RAI therapy in a subset of DTC.
KNOWN PATHWAYS DOWNREGULATING THE EXPRESSION OF NIS
MAPK/ERK pathway
The MAPK pathway has been well recognized and established in the regulation of cell proliferation, dedifferentiation, and survival [282930], particularly for PTC. Among the signal molecules of this pathway, BRAF mutations and the rearranged during transfection (RET)/PTC rearrangement are frequently detected in PTC, which often exhibit as exclusive mutations in such patients [53132]. The BRAFV600E mutation, as a prominently prevalent oncogene, is critical for the initiation and/or progression of PTC through aberrantly activating the MAPK signaling pathway, which can downregulate the expression of thyroid iodide-handling genes, especially NIS, and thus induce the dedifferentiation of PTC [2833343536]. Increasing evidence has demonstrated a strong association between the BRAFV600E mutation and the loss of RAI-avidity in PTC [37383940], which could provide a reasonable explanation for the failure of RAI therapy in BRAFV600E-mutant PTC.
As an important epigenetic event, histone acetylation plays a fundamental role in the regulation of gene transcription [4142]. Through aberrant activation of the MAPK pathway, histone acetylation at the promoter of the gene encoding NIS could be downregulated, which is considered as one of the key molecular events involving the aberrant silencing of thyroid iodide-handling genes [543444546]. Meanwhile, the BRAFV600E mutation can also upregulate the expression of tumor-promoting genes (e.g., vascular endothelial growth factor A [VEGFA], mesenchymal to epithelial transition factor [MET], TGFβ1, and thrombospondin 1 [TSP1]) [934474849] and downregulate the expression of tumor suppressor genes (e.g., tissue inhibitor of metalloproteinases 3 [TIMP3], solute carrier family 5 member 8 [SLC5A8], and death-associated protein kinase 1 [DAPK1]) [50], which are important constituents of the tumor microenvironment. It is noteworthy that the autocrine TGFβ loop could play a role in aberrant NIS expression. Riesco-Eizaguirre et al. [9] and Costamagna et al. [16] reported that the BRAFV600E mutation could induce the secretion of TGFβ, which subsequently stimulated SMAD3 and impaired PAX8, causing a decrease of NIS expression. As this process was independent of the MAPK pathway, their result undoubtedly indicates that TGFβ could be considered as a candidate therapeutic target for the restoration of NIS expression in patients with advanced DTC (Fig. 2).
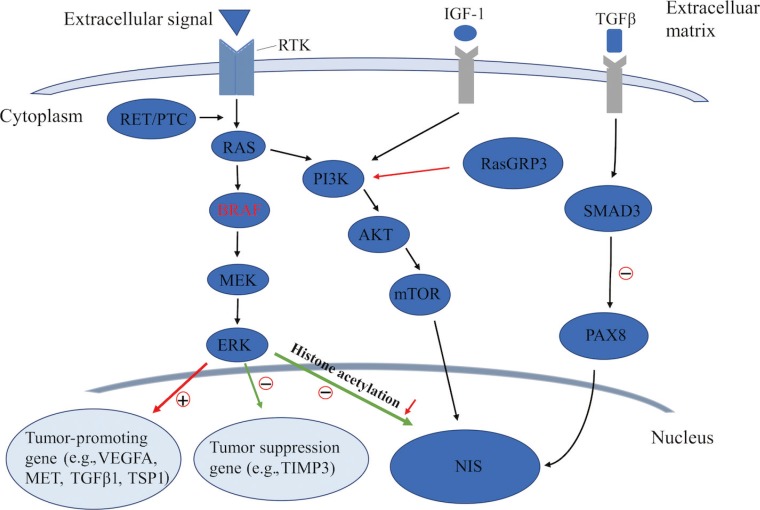
Known pathways involved in the regulation of sodium iodide symporter (NIS) in thyroid cancer. RTK, receptor tyrosine kinase; IGF-1, insulin-like growth factor-1; TGFβ, transforming growth factor β; PTC, papillary thyroid cancer; PI3K, phosphoinositide 3-kinase; RasGRP3, Ras guanyl releasing protein 3; PAX8, paired box gene-8; MEK, mitogen-activated extracellular signal-regulated kinase; ERK, extracellular regulated protein kinase; mTOR, mechanistic target of rapamycin; VEGFA, vascular endothelial growth factor A; MET, mesenchymal to epithelial transition factor; TSP1, thrombospondin 1; TIMP3, tissue inhibitor of metalloproteinases 3.
It was recently recognized that telomerase reverse transcriptase (TERT) promoter (TERTp) mutations are particularly prevalent in aggressive thyroid cancers, especially BRAFV600E-mutant PTC, but are virtually undetectable in benign thyroid neoplasms [5152]. TERTp mutations were demonstrated to be associated with aggressive tumor behavior and poor prognosis in thyroid cancer [535455], and were also observed to be correlated with the reduction of RAI uptake in distant metastatic lesions of PTC [56], which suggested that TERTp mutations may play a role in the dedifferentiation of thyroid cancer. It was also reported that the duet alternation of BRAFV600E and TERTp mutations have a robust synergistic effect on the progression and poor clinical outcomes of PTC. Recently, Liu et al. [57] revealed that the BRAFV600E/MAPK pathway could phosphorylate and activate fructooligosaccharide, which in turn acted as a transcription factor to activate the GA binding protein transcription factor subunit beta (GABPB) promoter, increasing GABPB expression and leading to the formation of the GA binding protein transcription factor subunit alpha (GABPA)-GABPB complex, thus activating the mutant TERT promoter and upregulating TERT expression. The discovery of TERTp mutations and the genetic duet of coexisting mutations could afford promising molecular targets for the salvage therapy of RAIR-PTC.
Concerning the RET/PTC rearrangement, evidence about its impact on the dedifferentiation of DTC remains limited. Trapasso et al. [58] and Wang et al. [59] reported that alternation of RET/PTC in the thyroid cell line could decrease the expression of thyroid differentiation markers, such as TSH receptor, TPO, NIS, and thyroglobulin. Furthermore, exogenous RET/PTC could significantly suppress the expression of PAX8 and the activity of PKA, leading to reduced NIS expression [858].
PI3K/AKT pathway
In addition to the MAPK pathway, the PI3K/AKT pathway also plays a fundamental role in controlling both cell proliferation and differentiation in DTC. In recent years, several genetic alterations activating the PI3K pathway have been identified in thyroid cancer, which was also shown to downregulate iodide-handling genes in thyroid cells [560]. Song et al. [61] recently reported that, also by this pathway, mutation of Ras guanyl releasing protein 3 (RasGRP3) decreased the expression of NIS and the TSH receptor in metastases of RAIR-DTC. Furthermore, the PI3K pathway could be activated by multiple growth factors such as insulin/insulin-like growth factor-1 (IGF-1) and epidermal growth factor [62]. Garcia and Santisteban [63] reported that IGF-1 could inhibit TSH-dependent NIS expression and reduce the iodide uptake in fisher rat thyroid cell line-5 (FRTL-5) rat thyroid cells through activating the PI3K/AKT pathway (Fig. 2). It has been shown that inhibition of the PI3K pathway by LY294002 could significantly increase the expression of NIS mRNA in rat thyroid cells and PTC cells, which, from another aspect, indicated the important role of the PI3K pathway in regulating NIS-mediated iodide accumulation in thyroid cancer [64]. The serine-threonine protein kinase mechanistic target of rapamycin (mTOR), which is located downstream of the PI3K/AKT pathway, has been identified as a regulator of cellular metabolism and proliferation. Souza et al. [65] reported that mTOR inhibition not only regulated cell survival, but also increased RAI uptake in both in vitro and in vivo studies.
REDIFFERENTIATION THERAPY FOR RAIR-DTC
During the past decade, although great progress has been achieved in the treatment of RAIR-DTC with the application of multi-kinase inhibitors (MKIs), therapeutic options for these patients are still limited. As RAI resistance is predominantly due to the dedifferentiation of DTC, redifferentiation therapy followed by RAI therapy undoubtedly is a promising alternative option for RAIR-DTC patients. Several agents, including retinoic acid (RA) [66], peroxisome proliferator-activated receptor gamma (PPARγ) agonists [67], and histone deacetylase (HDAC) inhibitors [68], have been tried to modulate the NIS gene at the transcription level, but displayed limited clinical value in redifferentiation therapy for patients with RAIR-DTC. Recent studies using drugs that selectively inhibit the MAPK and PI3K pathways showed promising results for restoring the expression of the gene encoding NIS and improving the response to RAI therapy in RAIR-DTC, such as mitogen-activated extracellular signal-regulated kinase (MEK)/RAF inhibitors [6970], PI3K/mTOR inhibitors [64] and receptor tyrosine kinase (RTK) inhibitors [71], which were used to restore NIS expression by suppressing the signaling pathways in such patients.
Agents modulating NIS at the gene transcriptional level
RA is a biologically active metabolite of vitamin A that play key roles in cell differentiation and proliferation. RA has been used for redifferentiation treatment of thyroid cancer, exerting its effects via retinoid receptors, RA receptors (RAR), or retinoid X receptors (RXR). Studies have shown that NIS mRNA expression was upregulated by RA stimulation in human follicular thyroid carcinoma cell lines [72]. Several early small cohort studies showed that 40% to 50% of patients with RAIR-DTC experienced renewed RAI uptake after RA treatment [737475]. However, such promising results could not be repeated by subsequent studies, which disappointingly indicated that only 6% to 20% of patients showed increased RAI uptake after RA administration [667677]. A recent open-label, non-randomized phase II trial reported even more disappointing results, with only one patient (1/16) showing increased RAI uptake after RA administration [66].
The PPARγ is a nuclear receptor which can regulate the proliferation and redifferentiation of the cell through forming a heterodimer with RXR [7879]. Pieces of evidence have shown that the rosiglitazone, a PPARγ agonist agent for the redifferentiation treatment of thyroid cancer, increased NIS mRNA expression and RAI uptake in thyroid cells [8081]. However, the results of a phase II clinical trial were disappointing, as 25% (5/20) of the patients displayed positive RAI uptake after rosiglitazone treatment, but no clinical response in long-term follow-up [67].
HDAC is an enzyme that deacetylates histones, which could silence the expression of NIS in thyroid cancer [5]. HDAC inhibitors were found to increase the expression of NIS mRNA at the epigenetic level in an in vitro study [82]. Preclinical studies have shown renewed RAI uptake after treatment with various HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA), depsipeptide (romidepsin), and valproic acid. Nevertheless, their independent significance in clinical trials was limited. Kelly et al. [83] reported that only one (1/3) advanced thyroid cancer patient showed increased RAI uptake after SAHA administration. In a phase I clinical trial of romidepsin, no positive RAI uptake was detected by RAI scintigraphy among 11 enrolled RAIR-DTC patients [84]. Furthermore, the results of a phase II clinical trial using romidepsin showed that only two patients experienced increased RAI uptake and no major response was observed in 20 patients with RAIR-DTC [68]. Nilubol et al. [85] performed a phase II clinical trial to evaluate the effect of valproic acid, without renewed RAI uptake.
MAPK inhibitors
The independent administration of retinoids, romidepsin, and rosiglitazone was shown to have limited clinical application in redifferentiation therapy for RAIR-DTC. With the development of molecular and cellular biology, multiple novel targets have been revealed, which could afford new options for the redifferentiation therapy of RAIR-DTC. Evidence has shown that agents selectively inhibiting the MAPK pathway, such as BRAF or MEK inhibitors, could induce thyroid gene expression and restore RAI uptake in thyroid cancer cells [4386]. The results of a clinical trial performed by Ho et al. [69] revealed that, after treatment with selumetinib, a selective MEK inhibitor, 124I uptake was increased in RAIR-PTC patients, which indicated the restoration of NIS expression in these patients. Meanwhile, it was interesting that the efficacy of selumetinib was better in patients with an NRAS mutation than those with the BRAFV600E mutation [69]. Furthermore, the prior phase II clinical trial of selumetinib seemed to suggest that BRAFV600E mutant patients exhibited longer median progression-free survival than patients with BRAF wild-type tumors [87]. This suggested a potential relationship between the therapeutic efficacy of selumetinib and genetic alterations. However, it was disappointing that the subsequent phase III trial of selumetinib failed to reconfirm its effect in the restoration of RAI uptake, which suggested that further studies are needed to explore the efficacy of MEK inhibitors in the redifferentiation of RAIR-DTC.
Another phase II clinical trial showed that dabrafenib, a BRAF inhibitor, increased RAI uptake in 60% (6/10) of BRAF-mutant RAIR-DTC patients, with two of the six achieving partial response after subsequent RAI therapy [88], which surpassed selumetinib in the treatment of BRAF-mutated RAIR-DTC. Vemurafenib, another BRAF inhibitor, could also restore RAI uptake in a subset of BRAF-mutant RAIR-DTC patients, which was likely due to the upregulation of thyroid-specific gene expression via inhibition of the MAPK pathway [70]. Earlier studies showed that HDAC inhibitors (such as SAHA) alone could induce the expression of NIS and faint RAI uptake in thyroid cancer cells [8990]; gratifyingly, a recent study reported that combined treatment with an HDAC inhibitor and a MAPK inhibitor (dabrafenib and selumetinib) showed a robust redifferentiation effect in BRAFV600E-mutated thyroid cancer cells [91]. This result suggests that the combined administration of HDAC and/or MAPK inhibitors might be a promising choice to improve the efficacy of redifferentiation therapy in RAIR-DTC patients. Furthermore, Nagarajah et al. [92] reported that extracellular regulated protein kinase (ERK) inhibitors could significantly increase the accumulation of 124I in BRAFV600E-mutant PTC cells, indicating that ERK inhibitors may be a potential candidate for redifferentiation therapy in BRAFV600E-mutant PTC.
PI3K inhibitors
As mentioned above, the aberrant activation of the PI3K/AKT/mTOR pathway could downregulate NIS expression [5], indicating that this pathway may be a potential therapeutic target for the redifferentiation therapy of RAIR-DTC. LY294002, a PI3K inhibitor, significantly upregulated the expression of NIS mRNA and improved iodide uptake through induction of PAX8 in DTC cell lines [6493]. Moreover, the inhibition of AKT also exhibited an increase of iodide uptake by mediating the expression of NIS in thyroid cells [93]. Plantinga et al. [94] reported that an mTOR inhibitor could induce iodine uptake through increasing thyroid transcription factor 1 (TTF1) expression in an in vitro study. Nevertheless, findings have not been reported from several in vivo studies that were initiated to evaluate the changes of iodine uptake by suppressing the PI3K pathway in RAIR-DTC patients. Hence, further studies might be necessary to elucidate the impact of PI3K/AKT/mTOR pathway inhibitors on the redifferentiation of RAIR-DTC.
RTK inhibitors
RTKs, such as vascular endothelial growth factor receptor (VEGFR), RET, platelet-derived growth factor receptors (PDGFRs), and human epidermal growth factor receptor (HER), are also crucial molecules inducing the dedifferentiation of DTC. By aberrantly causing the MAPK pathway to rebound, RTKs, such as HER3, could also lead to resistance to MKIs in RAIR-DTC patients [95]. Agents targeting RTKs have been recently investigated for the redifferentiation and salvage therapy of MKI-resistant RAIR-DTC. Cheng et al. [71] reported that combination therapy of BRAF/MEK inhibitors (dabrafenib/selumetinib) with an HER inhibitor (lapatinib) could upregulate NIS expression and suppress the MAPK pathway without a rebound phenomenon, and the corresponding phase I trial is now underway. The above-mentioned results may afford us a new prospective for the redifferentiation therapy of RAIR-DTC, as well as alternative salvage therapy for MKI-resistant advanced thyroid cancer.
CONCLUSIONS
The expression of NIS allows RAI therapy to be a highly effective management strategy for DTC, especially in metastatic DTC patients. Genetic alternations could reduce the expression of NIS and lead to the dedifferentiation of DTC, mainly through activation of the MAPK and PI3K pathways in thyroid cancer, causing a RAIR state that represents a life-threatening clinical situation. Efforts have been made to restore NIS expression and enhance RAI avidity in RAIR-DTC. Agents for dedifferentiation therapy at the transcriptional level have yielded limited clinical impact in clinical trials. Kinase inhibitors targeting the MAPK or PI3K pathways have shown promising effects in redifferentiation therapy and shed light on future combination therapy between either kinase inhibitors with different targets or kinase inhibitors and RAI in the management of RAIR-DTC.
ACKNOWLEDGMENTS
This review was supported by the National Natural Sciences Foundation of China (81771875 and 81571714), the Medicine and Technology Innovation Project of the Chinese Academy of Medical Science (grant number 2016-12M-2-006), and the Thyroid Study Group Project of the Asia Oceania Research Initiative Network (AORIN).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.