Characteristics of Acromegaly in Korea with a Literature Review
Article information
Abstract
Acromegaly is a slowly progressive disease caused by excessive growth hormone (GH), which is related to a GH secreting pituitary tumor in most cases. Herein, we describe the epidemiology, clinical characteristics, and treatment of acromegaly in Korea with a literature review. The average annual incidence of acromegaly in Korea was 3.9 cases per million people, which was within the range of previous Western studies. The primary treatment for acromegaly was also transsphenoidal adenomectomy, which accounted for 90.4% of patients whose primary therapeutic options were known. The overall surgical remission rates were 89%, 87%, 64%, 70%, and 50% for modified Hardy classification I, II, IIIA, IIIB, and IV, respectively. An updated and larger study regarding the treatment outcome of medical/radiotherapy in Korean acromegalic patients is needed.
INTRODUCTION
Acromegaly is a slow progressing disease caused by excessive growth hormone (GH), which is related to a GH secreting pituitary tumor in most cases. Patients with acromegaly generally exhibit acral enlargement, increased skin thickness, and facial bony deformities, including prognathism, nasal bone hypertrophy, and frontal bossing. More importantly, an approximate 30% mortality rate increase has been reported in patients with acromegaly. Particularly, cardiovascular disease represents the cause of death in 60%, respiratory disease in 25%, and malignancies in 15% of overall mortalities [1]. Recent meta-analysis showed approximately a 72% increase in all cause mortality in patients with acromegaly, compared with the general population, even after transsphenoidal adenomectomy (TSA) [2]. Therefore, early diagnosis and treatment to improve long-term morbidity and mortality, as well as relieve symptoms, are important.
Unfortunately, a clinical review of acromegaly data in Korea has not yet been performed. Herein, we described the epidemiology, clinical characteristics, and treatment of acromegaly in Korea with literature review.
EPIDEMIOLOGY IN KOREA
The prevalence of acromegaly has been estimated to be 40 to 70 cases per million with three to four cases per million of the annual incidence in Western countries [3]. However, recent studies have reported that the prevalence of acromegaly could be much higher, including 122 cases per million in a Belgium study [4] and 1,034 cases per million in a German study [5].
In Korea, the first nationwide survey regarding clinical characteristics of acromegaly was performed in 1994 [6]. Yang et al. [6] reported that 279 patients were newly diagnosed during the period from 1988 to 1992 with a rate of 1.4 cases per million, which is much lower than the estimated annual incidence of Western countries. However, patient and primary physician awareness, as well as improvement in medical care due to economic growth have increased the diagnostics rate of acromegaly. With this background, an updated version of a nationwide survey regarding acromegalic patients in Korea has been recently published. Kwon et al. [7] obtained all possible medical records of acromegalic patients collected by 74 secondary or tertiary medical institutes from 2003 to 2007. He reported that the prevalence of acromegaly was 27.9 cases per million in 2007 (Fig. 1). The average annual incidence of acromegaly was 3.9 cases per million, which was within the range of previous Western studies [7].

CLINICAL MANIFESTATIONS OF ACROMEGALY
The clinical manifestations of acromegaly are due to either to the consequences of GH hypersecretion or the tumor mass itself. GH hypersecretion leads to excessive growth, resulting in the classical features and medical comorbidities of acromegaly. The tumor mass effects can lead to headache, visual loss, and hypopituitarism. Unfortunately, there have been no reports on the clinical features of Korean acromegalic patients.
The typical features include signs of somatic overgrowth, including soft tissue swelling, enlarged hands and feet, arthralgias, and prognathism [8]. However, only a fraction of patients with acromegaly are actually diagnosed following presentation with a chief complaint attributable to acral overgrowth. In a review of 164 patients with acromegaly, only 58 (35%) presented because of a change in features [9]. In this study, 56 patients (34%) presented because of disturbances associated with acromegaly, including visual field defects, carpal tunnel syndrome, and headaches. The remaining 50 individuals had no complaints related directly to acromegaly, and were diagnosed when seeking medical attention for unrelated complaints. These data demonstrate the difficulties in detecting the disease, as it is relatively uncommon that patients present with complaints attributable to the more classical signs of acromegaly, including bony or soft tissue overgrowth. In addition, because the features of acromegaly progress in an insidious fashion, there is often a 7 to 10 year diagnosis delay after the estimated onset of symptoms [10].
Besides the mass effect, chronic GH and insulin-like growth factor 1 (IGF-1) hypersecretion can lead to a myriad of soft tissue and bone overgrowth manifestations, medical comorbidities, and accompanying clinical features. They include stimulation of growth of many tissues, such as skin, connective tissue, cartilage, bone, viscera, and epithelial tissues. Concerning the mortality of acromegaly, many studies have been focused on the development of cardiovascular disease and neoplasms.
Hypertension is detected in up to 40% of individuals, and is presumably secondary to an increase in plasma volume along with an increase in sodium retention [11]. Suppressed plasma rennin activity and aldosterone concentrations in acromegaly are consistent with a primary increase in total body sodium. The presence of insulin resistance, a hyperdynamic cardiomyopathy with increased cardiac output, as well as morphological alterations and/or endothelial dysfunction may also contribute to the hypertension [12]. Cardiovascular abnormalities include hypertension, left ventricular hypertrophy, and cardiomyopathy. Interestingly, acromegaly may be associated with an increase in carotid artery intima-media thickness, though the prevalence of atherosclerosis may be similar to that of the general population. This finding suggests that GH and/or IGF-1 may limit the progression of atherosclerosis, despite the coexistence of hypertension and glucose intolerance. An acromegalic cardiomyopathy, characterized by biventricular cardiac hypertrophy, is a hallmark finding in patients with acromegaly. Both age and disease duration correlate with the presence and degree of the hypertrophy [13]. The cardiomyopathy may progress with signs of diastolic dysfunction and/or insufficient systolic performance early in the disease process, to systolic dysfunction at rest, and, rarely with advanced disease, overt heart failure [14]. Cardiac valve dysfunction may be detected in acromegaly patients; arrhythmias, including atrial fibrillation, supraventricular tachycardia, and bundle branch blocks, may be detected as well [15].
An increased risk of cancer, particularly colonic, with acromegaly has been suggested largely by retrospective studies, though this finding is controversial [8,16]. In one study, the overall cancer mortality rate was not increased, but the colon cancer mortality rate (standardized mortality ratio, 2.47) was higher than expected for the general population [17]. In addition, many, but not all, studies have reported that colonic polyps are more prevalent in acromegalic patients than controls [18]. These findings are highly suggestive of a connection between GH hypersecretion and neoplasm growth, particularly of the colon.
TREATMENT OUTCOME OF ACROMEGALY IN KOREA
The treatment of acromegaly focuses on normalization of GH/IGF-1 secretion, which is the most important factor required for reversing the observed increased morbidity and mortality. TSA is generally the first line treatment for acromegaly. When surgery fails to achieve control of GH/IGF-1 hypersecretion, medical treatment with a somatostatin analogue is preferred to radiotherapy.
Although surgery is the most rapid way of reducing tumor volume and GH/IGF-1, the overall surgical cure rate has been reported only as 40% to 70%, depending on the size of the tumor (microadenoma, 70% to 90%; macroadenoma, less than 50%), preoperative GH concentrations, and the surgeon's experience [19-21].
In Korea, according to the report by Kwon et al. [7], among 1,137 patients whose primary therapeutic options were known, TSA was also the primary treatment in 1,028 patients (90.4%). The remission rate at 3 months after TSA was 62.8% and 44.1% for microadenoma and macroadenoma, respectively. Among 344 patients who were not in remission three months after TSA from 2003 to 2007 and whose medical treatment histories were collected, 157 patients (45.6%) received medical treatment including somatostatin analogue or dopamine agonist.
A retrospective study of 282 patients with GH-secreting pituitary tumors who underwent TSA by a single experienced neurosurgeon (S.H.K.) over a 17-year period was recently published [22]. The study showed the overall surgical remission rates were 89%, 87%, 64%, 70%, and 50% for modified Hardy classifications I, II, IIIA, IIIB, and IV, respectively (Table 1). Ku et al. [22] also suggested that extensive surgical resection could achieve higher remission rates and lower recurrence rates without aggravating postoperative hypopituitarism.
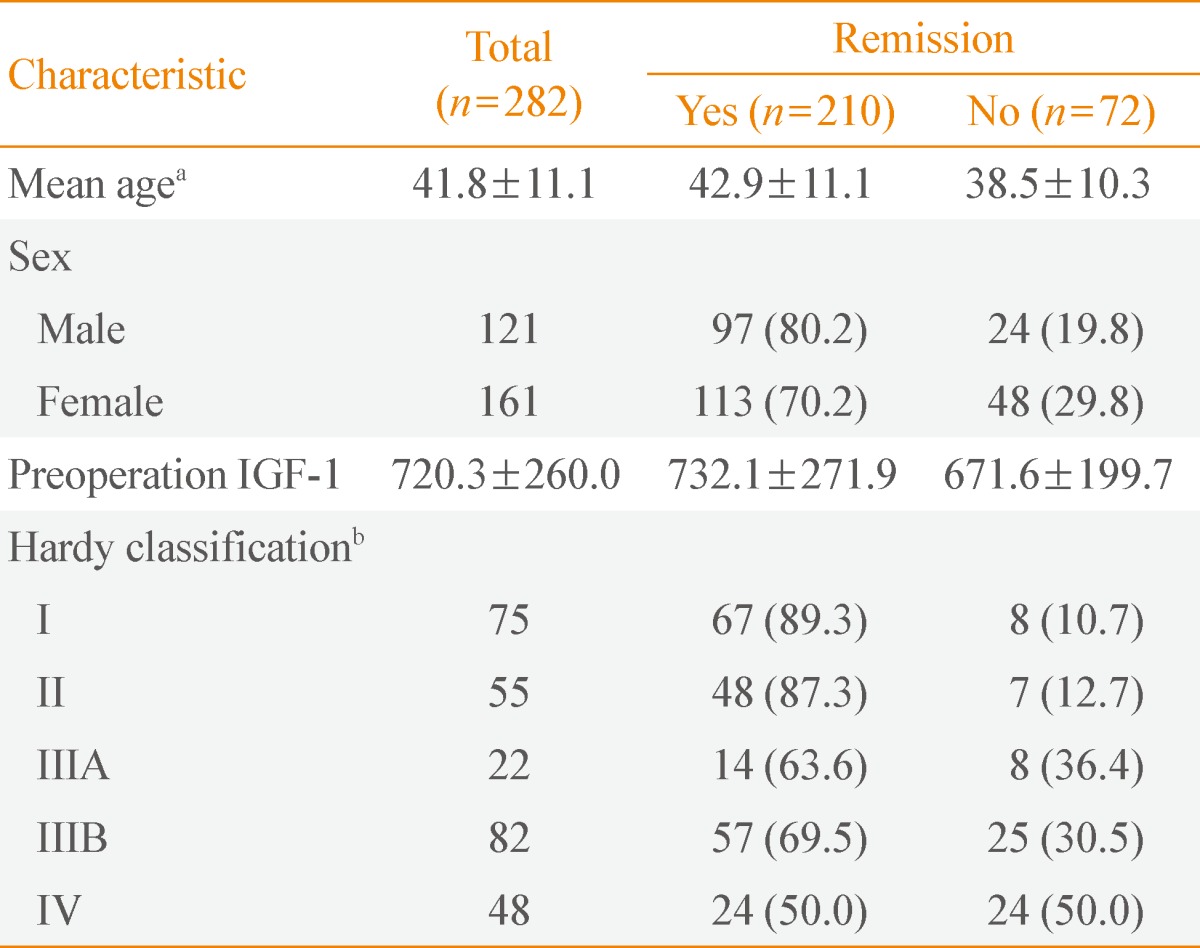
Clinical Characteristics according to Surgical Outcome in Patients with Acromegaly
Medical therapy for acromegaly is usually indicated after unsuccessful TSA and while awaiting the effect of radiotherapy [1]. Dopamine agonists and somatostatin analogues are available in Korea. Bromocriptine has been traditionally used in the past to treat acromegaly. However, cabergoline has been shown to be more effective and have less side effects than bromocriptine. A meta-analysis showed cabergoline monotherapy can normalize IGF-1 levels in one third of acromegalic patients. When a somatostatin analog fails to control acromegaly, cabergoline cotreatment was shown to normalize IGF-1 in about 50% cases, even in patients with normoprolactinemia [23]. Somatostatin analogues, including octreotide and lanreotide, suppress GH secretion by binding to somatostatin receptors, SST2 and 5. These drugs normalize IGF-1 levels in 50% to 80% patients and reduce the tumor volume in 20% to 70% of patients [21]. Unfortunately, there has been no data generated regarding the outcome of medical treatment in Korean acromegalic patients.
Radiotherapy can reduce tumor volume and GH/IGF-1 hypersecretion, but the onset of action is delayed and hypopituitarism usually develops [21]. External beam radiotherapy (EBRT) normalized IGF-1 level in about 36% of patients [24]. Stereotactic radiosurgery allows the administration of a very large radiation dose to an extremely defined area and induces a decrease in GH/IGF-1 levels more effectively and faster than conventional EBRT. Approximately 58% of patients treated with stereotactic radiosurgery achieved normalization of GH/IGF-1 level [25]. However, the role of radiotherapy has become more and more controversial due to the improvement of surgical techniques and availability of newer, more effective drugs [1,24].
In Korea, the local control rate of radiotherapy including EBRT and stereotactic radiosurgery of pituitary tumor were as reported 66% and 92%, respectively, in recent data. However, the numbers of acromegaly patients enrolled in those studies were very small (n=9 and n=6) [26,27].
An updated and larger study regarding the medical/radiotherapy treatment outcome of acromegaly is needed.
CONCLUSIONS
The average annual incidence of acromegaly in Korea was 3.9 cases per million, which was within the range of previous Western studies. The primary treatment of acromegaly was also TSA, which accounts for 90.4% of patients whose primary therapeutic options were known. Overall surgical remission rates were above 85% in intrasellar adenoma, however, only 50% to 70% in extrasellar adenoma. An updated and larger study regarding the medical/radiotherapy treatment outcome in Korean acromegalic patients is needed.
Notes
No potential conflict of interest relevant to this article was reported.