Articles
- Page Path
- HOME > Endocrinol Metab > Volume 36(2); 2021 > Article
-
Review ArticleRole of PCSK9 Inhibitors in Patients with Familial Hypercholesterolemia
-
Brian Tomlinson
 , Nivritti Gajanan Patil, Manson Fok, Christopher Wai Kei Lam
, Nivritti Gajanan Patil, Manson Fok, Christopher Wai Kei Lam -
Endocrinology and Metabolism 2021;36(2):279-295.
DOI: https://doi.org/10.3803/EnM.2021.964
Published online: April 19, 2021
Faculty of Medicine, Macau University of Science and Technology, Macau, China
- Corresponding authors: Brian Tomlinson. Faculty of Medicine, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau 999078, China, Tel: +853-8897-2415, Fax: +853-2882-5057, E-mail: btomlinson@must.edu.mo
- Christopher Wai Kei Lam. Faculty of Medicine, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau 999078, China, Tel: +853-8897-2636, Fax: +853-2882-5886, E-mail: wklam@must.edu.mo
Copyright © 2021 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Patients with familial hypercholesterolemia (FH) are at high or very high risk for cardiovascular disease. Those with heterozygous FH (HeFH) often do not reach low-density lipoprotein cholesterol (LDL-C) targets with statin and ezetimibe therapy, and those with homozygous FH (HoFH) usually require additional lipid-modifying therapies. Drugs that inhibit proprotein convertase subtilisin/kexin type 9 (PCSK9) offer a novel approach to reduce LDL-C. The monoclonal antibodies, alirocumab and evolocumab, given by subcutaneous injection every 2 or 4 weeks produce reductions in LDL-C of 50% to 60% in patients with HeFH, allowing many of them to achieve their LDL-C goals. Patients with HoFH show a reduced and more variable LDL-C response, which appears to depend on residual LDL receptor activity, and those with receptor-negative mutations may show no response. Inclisiran is a long-acting small interfering RNA therapeutic agent that inhibits the synthesis of PCSK9. Subcutaneous doses of 300 mg can reduce LDL-C by more than 50% for at least 6 months and the responses in HeFH and HoFH patients are similar to those achieved with monoclonal antibodies. These PCSK9 inhibitors are generally well tolerated and they provide a new opportunity for effective treatment for the majority of patients with FH.
- Familial hypercholesterolemia (FH) is a genetic autosomal codominant condition causing high levels of low-density lipoprotein cholesterol (LDL-C), which predispose individuals to premature atherosclerotic cardiovascular disease (ASCVD) and especially coronary heart disease (CHD) [1]. Heterozygous FH (HeFH) was originally thought to occur in about 1/500 people, but recent estimates suggest the prevalence is about 1/200 to 1/300 in most populations [2]. The phenotype of HeFH is quite variable and many people with the condition are not aware of it unless they undergo genetic testing or until they suffer an ischemic event [3,4].
- Patients with homozygous FH (HoFH) also exhibit a variable phenotype, but are generally easier to recognize as they usually present the clinical features of tendon xanthomas and cutaneous xanthomas in childhood and typically develop very early CHD unless suitable interventions are implemented from an early age [5]. The prevalence of autosomal dominant HoFH in the Netherlands was estimated to be about 1/300,000 in a report in 2015 [6].
INTRODUCTION
- Clinical criteria have often been used for the diagnosis of FH [7–9]. These include the Simon Broome Register Group criteria, the Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria, and the Dutch Lipid Clinic Network criteria. Recently, the American Heart Association (AHA) developed revised criteria [10]. The Japan Atherosclerosis Society has also developed their own criteria, which include measurements of the Achilles tendon thickness as an objective assessment of tendon involvement [11].
- It is desirable to confirm the diagnosis of FH with genetic testing to optimize CHD risk assessment and facilitate cascade screening [12]. Monogenic FH most often occurs due to missense loss-of-function (LOF) mutations in the low-density lipoprotein receptor gene (LDLR) and over 2,300 different LDLR mutations have been identified and listed in the ClinVar database in 2018 [13]. The next most commonly affected gene is APOB, which encodes apolipoprotein B (apoB); mutations in this gene are often associated with a somewhat less severe phenotype that is also referred to as familial defective apoB [3,10]. The frequency of different mutations varies in different parts of the world and some common mutations in the LDLR and APOB genes have been described in Asian countries [14,15]. Common mutations occur in some regions as a founder effect causing an increased local population prevalence of FH [16].
- An autosomal recessive form of FH caused by LOF mutations in the LDL receptor adaptor protein 1 gene (LDLRAP) was described in 2003 [17]. The clinical phenotype varies but may be less severe than that of classic dominant HoFH caused by true homozygous or compound heterozygous LDLR mutations. More recently, researchers have identified another autosomal recessive genetic cause involving lysosomal acid lipase deficiency with mutations in the lipase A gene (LIPA), which results in a wide spectrum of phenotypes including cholesterol ester storage disease and Wolman disease [18,19].
- Proprotein convertase subtilisin/kexin type 9 (PCSK9) was discovered as the product of the third gene found to cause autosomal dominant FH due to gain-of-function (GOF) mutations in PCSK9 [20]. PCSK9 was recognized to be the same as the secretory proprotein convertase, which had been called neural apoptosis-regulated convertase 1 (NARC-1) [21]. This discovery provided a new target for treatment to reduce LDL-C levels. This is particularly relevant for patients with FH, as many of those with HeFH do not reach LDL-C targets with the maximum tolerated statin therapy or even with the addition of ezetimibe [22].
- Many people thought to have FH may have a high polygenic score rather than monogenic dominant or recessive conditions. In a cohort of 313 individuals with severe hypercholesterolemia, defined as LDL-C >5.0 mmol/L (>194 mg/dL), monogenic FH-causing mutations were present in 47.3% of individuals and monogenic mutations increased to 53.7% when copy number variations were included; the percentage with a genetic component further increased to 67.1% when individuals with extreme polygenic scores were included [23]. Polygenic contributions to LDL-C also explain some of the heterogeneity in clinical presentation and ASCVD risk for individuals with FH [24].
DIAGNOSIS OF FAMILIAL HYPERCHOLESTEROLEMIA
- In the 2019 European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) guidelines, patients with FH without additional risk factors are considered high risk and the LDL-C treatment goal is <1.8 mmol/L (<70 mg/dL), along with a reduction of ≥50% from baseline, whereas those with ASCVD or another major risk factor are considered very high risk and the LDL-C treatment goal is <1.4 mmol/L (<55 mg/dL) [25]. The 2018 Korean guidelines for the management of dyslipidemia recommend a target LDL-C <100 mg/dL for FH patients without CHD or other major risk factors and <70 mg/dL when CHD or other major risk factors are present [9].
- The risk for ASCVD in FH is influenced by the usual cardiovascular risk factors including lipoprotein(a) (Lp(a)) [26]. Lp(a) levels are raised in FH and are an important predictor of cardiovascular disease independent of the type of LDL receptor mutation [27]. Marked elevation of Lp(a) may be present in 30% to 50% of patients with FH, and the cholesterol content of Lp(a) contributes to LDL-C measurements and may influence the LDL-C threshold diagnostic criteria for FH used in clinical algorithms [28]. Statins tend to increase Lp(a) [29]; therefore, having other treatments that reduce it may be an advantage and some FH patients may need specific treatment to target Lp(a).
- In a study of HeFH patients in the Netherlands, statin treatment reduced the risk of myocardial infarction to a level that was not significantly different from that of an age-matched sample from the general population [30]. A 20-year follow-up study of statin therapy in children in the Netherlands showed that early initiation of treatment in FH patients can considerably reduce the risk of ASCVD [31]. However, another study of HeFH patients in the Netherlands found that only about half of them reached a target LDL-C of <3 mmol/L and only 21% achieved a target of <2.5 mmol/L [22]. Many patients were not on maximum statin doses and only 54% of patients were on ezetimibe. The main reason for patients not reaching the LDL-C targets appeared to be inertia of the treating physician.
- In a prospective study of HeFH patients in Korea, the LDL-C target of <100 mg/dL was achieved by 28% of patients and 47% achieved a ≥50% LDL-C reduction at 12 months with high-intensity statin regimens or combination therapy; according to that study, pretreatment LDL-C was a major determinant of the response [32].
- Lipid-lowering therapy, predominantly with statins, has improved the prognosis of patients with HoFH [33], but they usually require additional lipid-modifying therapy such as lomitapide, and adjunctive lipoprotein apheresis is often utilized when available [5]. Having a highly effective treatment to inhibit PCSK9, such as the fully human monoclonal antibodies (mAbs), alirocumab and evolocumab, can enable many more patients with FH to achieve the recommended LDL-C goals [34,35].
GOALS AND LIPID-LOWERING TREATMENTS FOR FAMILIAL HYPERCHOLESTEROLEMIA
- Numerous studies have investigated the role of PCSK9 in lipid metabolism and the effects of genetic variations in PCSK9. Two nonsense mutations—c.426C>G (rs67608943, p.Y142X) and c.2037C>A (rs28362286, p.C679X) in PCSK9—were found to be common in African Americans and were associated with a 40% reduction in plasma levels of LDL-C [36]. Further studies showed that these nonsense mutations in PCSK9 were associated with a 28% reduction in mean LDL-C and an 88% reduction in the risk of CHD in African Americans, while the p.R46L (rs11591147, c.137G>T, p.Arg46Leu) variation in PCSK9 that was more common in white subjects was associated with a 15% reduction in LDL-C and with a 47% reduction in the risk of CHD over a 15-year interval [37].
- An analysis of three studies from Denmark also showed that the PCSK9 R46L variant was associated with a modest reduction of LDL-C levels and a more marked reduction of CHD risk compatible with a lifetime exposure to lower LDL-C [38]. The reported variants in PCSK9 present a spectrum of hypercholesterolemia or hypocholesterolemia phenotypes [39].
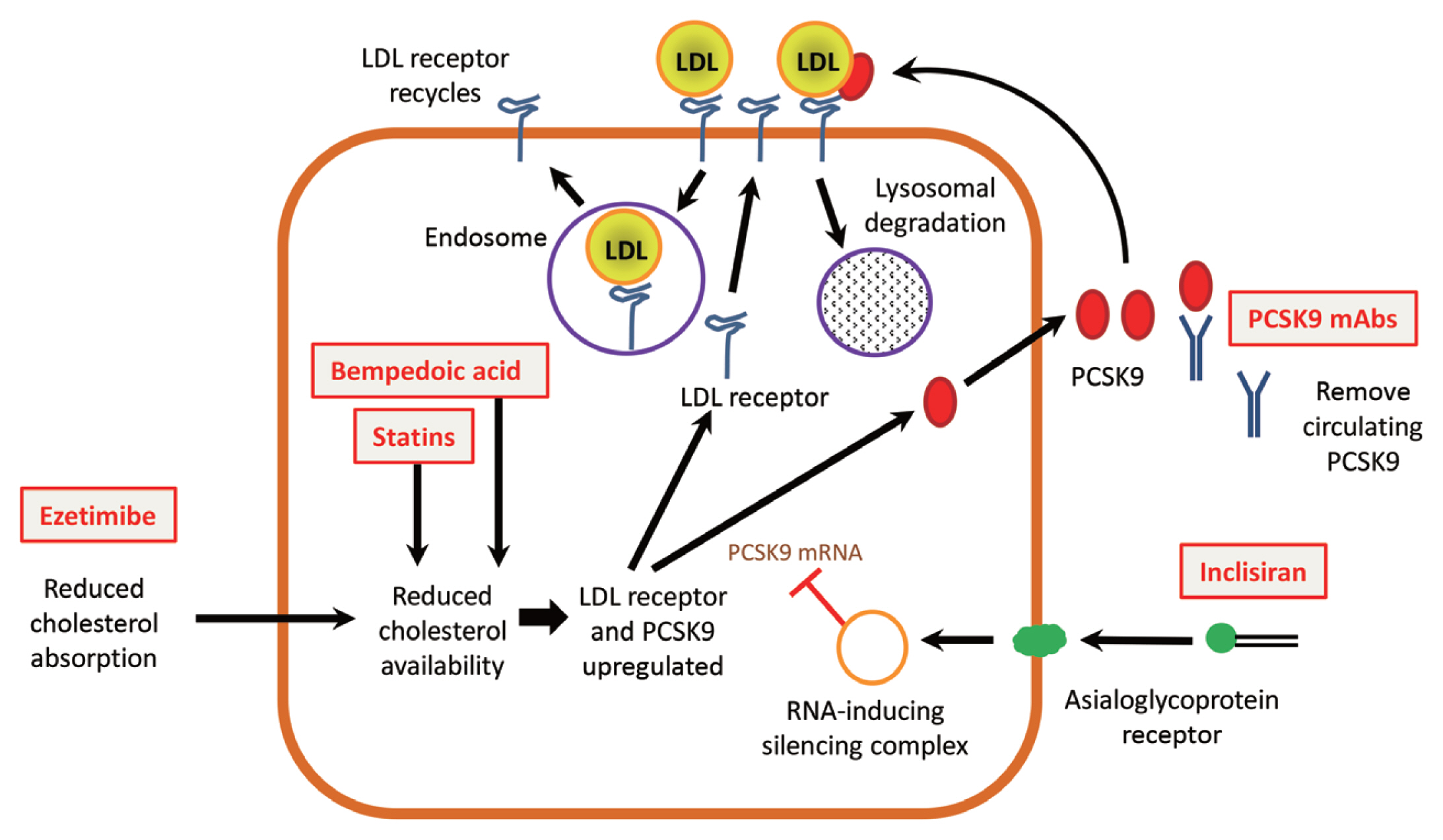
- It was shown in several studies that PCSK9 induces the degradation of the LDLR and related receptors by acting as a chaperone that binds the LDLR during receptor-mediated endocytosis and then directs it for lysosomal degradation (Fig. 1) [40–42]. The enzymatic activity of PCSK9 is not involved in the process. Statins upregulate PCSK9 by causing intracellular sterol depletion and increasing levels of the sterol-regulatory element binding protein-2 (SREBP-2), which activates transcription of PCSK9 as well as the LDLR [43–45]. When the dose of statin is increased, the increase in levels of PCSK9 attenuates the increased expression of the LDLR, which is thought to account for the limited additional LDL-C lowering effect of about 6% when statin doses are doubled [46].
- PCSK9 levels were elevated in untreated FH patients, particularly in those with HoFH, and high-dose statin therapy further increased PCSK9 levels in these patients [47]. Moreover, adding ezetimibe to statin resulted in additional increases in PCSK9 levels [48].
PROPROTEIN CONVERTASE SUBTILISIN/KEXIN TYPE 9
- The mAbs developed to target PCSK9 include alirocumab and evolocumab, which are fully human mAbs, and bococizumab which is a humanized mAb that might be expected to have some risk of immunogenicity [49]. Indeed, the development of bococizumab was discontinued when it was seen that there was an attenuation of LDL-C lowering over time as well as higher rates of injection-site reactions in the Studies of PCSK9 Inhibition and the Reduction of vascular Events (SPIRE) trials [50,51].
- In two phase II studies in which bococizumab was given intravenously every 4 weeks for 12 weeks in various doses to subjects with primary hypercholesterolemia on high doses of statin, 15% of subjects had genetic variants associated with FH and they responded in a similar way to subjects who did not carry these variants [52]. In the SPIRE program, 1,578 patients with FH were compared with 15,959 patients without FH selected for comparable lipid levels who had been randomized to bococizumab 150 mg subcutaneously (SC) every 2 weeks (Q2W) or to matching placebo and followed over a median period of 11.2 months [53]. The FH and non-FH patients both had reductions in LDL-C of 55% with bococizumab, but the proportion of patients developing antidrug antibodies was higher among those with FH than among those without FH (43% vs. 36%, P<0.001).
- Despite the attenuation of LDL-C lowering over time with bococizumab, the SPIRE-2 trial, which enrolled higher risk patients with LDL-C ≥100 mg/dL, showed a significant 21% reduction in major cardiovascular events over a median of 10 months illustrating the greater benefit of PCSK9 inhibition in higher-risk patients [50].
- Evolocumab
- Evolocumab is an immunoglobulin G 2 (IgG2)-isotype fully human mAb developed for use at doses of 140 mg SC Q2W or 420 mg SC monthly (QM), which have similar effects on LDL-C reduction [54,55]. It was approved by the U.S. Food and Drug Administration (FDA) in 2015 as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with HeFH or clinical ASCVD, who require additional lowering of LDL-C and as adjunct to diet and other LDL-lowering therapies (e.g., statins, ezetimibe, LDL apheresis) in patients with HoFH who require additional lowering of LDL-C. The effects of evolocumab in studies in patients with HeFH and HoFH are shown in Table 1.
- Evolocumab was tested in 167 patients with HeFH diagnosed by the Simon Broome criteria with LDL-C ≥2.6 mmol/L (100 mg/dL) despite statin therapy with or without ezetimibe in the phase 2 Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) study [56]. The HeFH patients were randomized to receive evolocumab 350 or 420 mg or placebo SC QM and at week 12. The mean±standard deviation (SD) LDL-C reduction was 42.7%±2.9% with 350 mg and 55.2%±2.9% with 420 mg evolocumab compared with an increase of 1.1%±2.9% with placebo (P<0.001 for both dose groups) (Table 1). There was some diminution in the LDL-C lowering effect between 2 and 4 weeks after giving evolocumab, even with the 420 mg dose; therefore, these changes at week 12 (i.e., 4 weeks after the last dose) may not be the maximum effect [57].
- In the phase 3 RUTHERFORD-2 study, 331 eligible patients meeting the clinical criteria for HeFH and fasting LDL-C ≥2.6 mmol/L on stable lipid-lowering therapy for at least 4 weeks, were randomized to evolocumab, given SC either 140 mg Q2W or 420 mg QM, or a matching placebo [58]. The reduction in mean LDL-C with evolocumab compared to placebo at the mean of weeks 10 and 12 was 60.2% (95% confidence interval [CI], 54.5% to 65.8%) in 111 subjects with the 140 mg dose Q2W and 65.6% (95% CI, 59.8% to 71.3%) in 110 subjects with the 420 mg dose QM. The reductions in LDL-C tended to be greater at week 10 than at week 12 with the 420 mg QM dose. The drug was well tolerated.
- In a small retrospective study of patients with hypercholesterolemia who were treated with evolocumab 140 mg SC Q2W, 32 patients with monogenic HeFH had similar reductions in LDL-C compared with seven patients with polygenic hypercholesterolemia (63.9%±16.0% and 67.7%±20.7%, respectively; no significant difference) [59].
- In the recent Trial Assessing Efficacy, Safety and Tolerability of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibition in Paediatric Subjects With Genetic Low-Density Lipoprotein (LDL) Disorders (HAUSER-RCT), 157 pediatric patients aged 10 to 17 years with HeFH diagnosed by genetic testing or clinical diagnostic criteria with LDL-C ≥130 mg/dL (3.4 mmol/L) on stable lipid-lowering treatment for at least 4 weeks were treated with evolocumab 420 mg or placebo QM for 24 weeks [60]. The mean±SD age of the patients was 13.7±2.4 years and the placebo-corrected reduction in LDL-C at 24 weeks was 38.3%, or 68.6 mg/dL (1.8 mmol/L) in absolute terms. The effect was somewhat greater at 22 weeks than at 24 weeks.
- Patients with HoFH with mutations in LDLR usually respond less well than those with HeFH to any treatment that involves the LDLR in the mechanism of action, such as statins and PCSK9 inhibitors. In the Trial Evaluating PCSK9 Antibody in Subjects With LDL Receptor Abnormalities (TESLA) pilot study, eight HoFH patients with LDLR-negative or LDLR-defective mutations on stable drug therapy were treated with open-label SC evolocumab 420 mg Q4W for at least 12 weeks, followed by 420 mg Q2W for an additional 12 weeks [61]. The two receptor-negative patients showed no reduction in LDL-C, whereas the receptor-defective patients showed mean±SD reductions in LDL-C of 19.3%±16% and 26.3%±20% with 4- and 2-week dosing, respectively, with a wide range of responses (4% to 48% with 2-week dosing).
- In the TESLA Part B phase 3 study, 50 eligible patients with HoFH who were on stable lipid-regulating therapy for at least 4 weeks and not receiving lipoprotein apheresis were randomized to treatment with SC evolocumab 420 mg or placebo Q4W for 12 weeks [62]. Of the 49 patients who received the study drug and completed the study, LDL-C was significantly reduced at 12 weeks with evolocumab by 30.9% (95% CI, 18.0% to 43.9%).
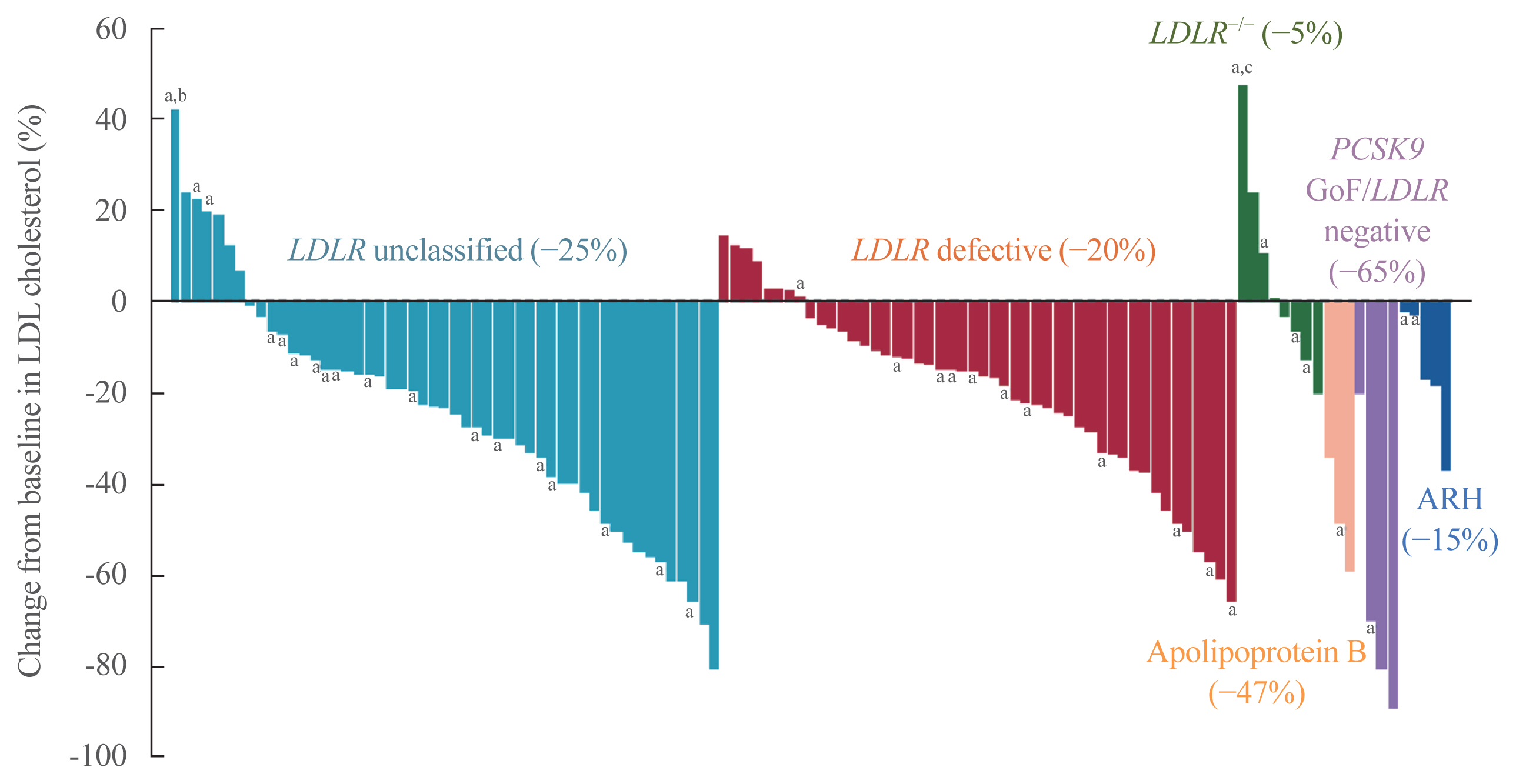
- An interim subset analysis of the open-label phase 3 Trial Assessing Long-Term Use of PCSK9 Inhibition in Subjects with Genetic LDL Disorders (TAUSSIG) study involved 106 patients with HoFH, including 34 who were receiving apheresis at study entry, with a mean follow-up of 1.7 years [63]. The patients received evolocumab 420 mg SC QM, or 420 mg SC Q2W if on apheresis, and the dose could be increased for patients not on apheresis to Q2W after 12 weeks. The mean LDL-C level decreased from baseline at week 12 by 20.6%±24.4% (mean absolute decrease, 1.50±1.92 mmol/L) and at week 48 by 23.3%±30.8%. In patients not on apheresis, there was an additional mean reduction in LDL-C of 8.3%±13.0% (mean absolute decrease, 0.77±1.38 mmol/L) when dosing was increased from QM to Q2W.
- There were considerable differences in responses between patients and according to mutation type and dosing regimen (Fig. 2). In the 47 receptor-defective patients not on apheresis, the mean±SD LDL-C reduction was 20.0%±19.6% at week 12 when they were being dosed QM, and 23.9%±21.9% at week 48, when they were up-titrated to dosing Q2W. The eight patients with LDLR negative mutations had a poor response, whereas the three patients with APOB mutations had a mean±SD 47.1%±12.2% reduction in LDL-C at week 12 and the four patients with a PCSK9 GOF mutation combined with an LDLR mutation had a mean±SD 64.8%±31.0% reduction in LDL-C at week 12. The reductions in LDL-C did not significantly differ in patients on apheresis or not on apheresis, and some of those on apheresis were able to reduce the frequency of the procedure or even discontinue it.
- In a final report from the TAUSSIG study, which included 106 patients with HoFH and 194 patients with severe HeFH on stable lipid-lowering therapy who received evolocumab for a median duration of 4.1 years at a dose of 420 mg QM or 420 mg Q2W, the mean±SD reduction in LDL-C from baseline to week 12 was 21.2%±25.0% in patients with HoFH and 54.9%±17.4% in those with severe HeFH; this reduction was sustained over the period of the study [64]. In 48 patients with HoFH who were up-titrated from evolocumab 420 mg QM to 420 mg Q2W, the mean reduction in LDL-C increased from 19.6% at week 12 to 29.7% after 12 weeks of 420 mg Q2W, and 16 of the 61 patients receiving apheresis at enrollment were able to discontinue apheresis. Evolocumab was well tolerated and no patients developed neutralizing antibodies.
- The Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) trial examined cardiovascular outcomes with evolocumab in high-risk patients with established stable cardiovascular disease [65]. There was a significant reduction in the primary composite cardiovascular endpoint with a hazard ratio of 0.85 (95% CI, 0.79 to 0.92; P<0.001) for patients treated with evolocumab compared with placebo over a median duration of follow-up of 2.2 years.
- In addition to reducing LDL-C, evolocumab decreases the levels of other atherogenic lipoproteins including Lp(a), which was reduced by median values of 24.7% and 21.7% with biweekly and QM doses of evolocumab, respectively, in an analysis of 10 clinical trials [66]. Changes in Lp(a) were quite variable, but were related to LDL-C reductions and appear to be mediated in part by increased LDLR uptake of Lp(a) particles.
- Alirocumab
- Alirocumab is a fully human mAb of the IgG1 isotype that binds with high affinity to PCSK9 [67,68]. It was approved by the FDA in 2015 as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with HeFH or clinical ASCVD, who require additional lowering of LDL-C. The LDL-C responses to alirocumab in studies in patients with HeFH and HoFH are shown in Table 2.
- A phase 1 study was conducted in patients with hypercholesterolemia, including some with HeFH [68]. The patients with HeFH and some of the subjects with non-FH were on atorvastatin. Doses of 50, 100, and 150 mg of alirocumab were given SC at day 1, 29, and 43. There was a dose-dependent maximum reduction in LDL-C seen at 8 to 15 days after the first dose, with mean reductions of 39.2%, 53.7%, and 61.0%, respectively, for the three doses. The response was similar in patients with and without concomitant atorvastatin treatment and in those with and without HeFH. The duration of LDL-C reduction was also dose-dependent and persisted longer in patients who were not taking atorvastatin. Those findings are consistent with statin treatment resulting in increased synthesis of PCSK9, which would reduce the duration of the effect of alirocumab by increasing the target-mediated clearance of the antibody bound to PCSK9 [69].
- In a phase 2 study in 77 patients with HeFH and LDL-C concentrations ≥2.6 mmol/L (100 mg/dL) who were on a stable statin dose, with or without additional ezetimibe, the patients were randomized to SC alirocumab 150, 200, or 300 mg Q4W alternating with placebo Q4W, or to alirocumab 150 mg or placebo Q2W, so they all received injections Q2W [70]. The mean reductions in LDL-C from baseline at week 12 for the different alirocumab doses were 28.9%, 31.5%, 42.5%, and 67.9%, respectively, compared with 10.7% with placebo. The reductions in LDL-C at 2 weeks after the doses given every 4 weeks were greater than those at 4 weeks, showing that the LDL-C reduction was not fully maintained for 4 weeks even with the 300 mg Q4W dose, whereas the 150 mg Q2W dose had a consistent effect that reached a maximum after 3 doses [70].
- In a 3-year open-label treatment extension of the 12-week double-blind phase 2 study in HeFH patients, 58 patients received alirocumab 150 mg Q2W, and the mean±SD reduction in LDL-C was 65.4%±21.1% at week 24 and 56.0%±23.8% at week 148 [71]. Over the course of the study, 12 patients (20.7%) experienced a serious treatment-emergent adverse event and injection site reactions occurred in 21 (36.2%) patients; nonetheless, alirocumab was considered to be generally well-tolerated in the long term [71].
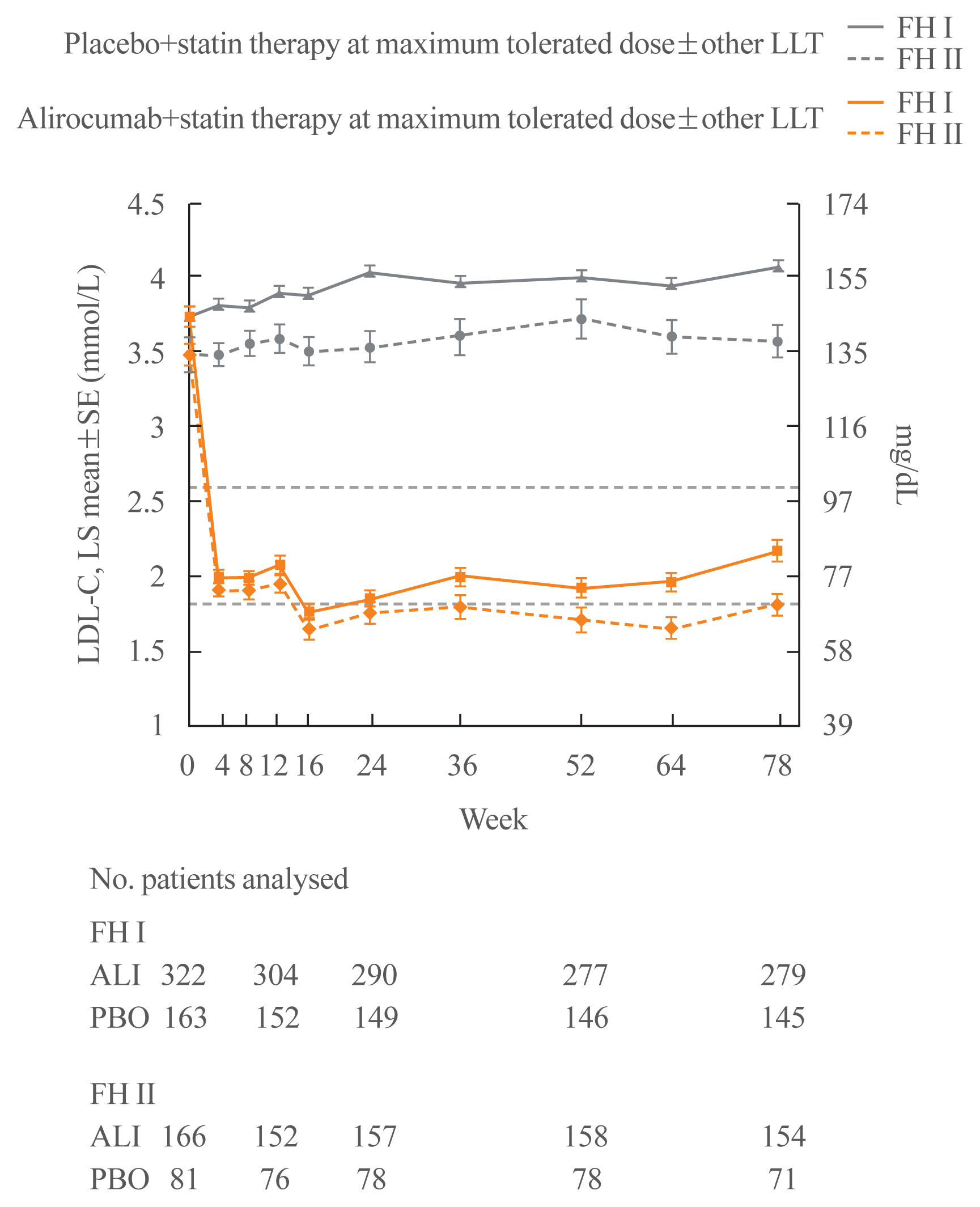
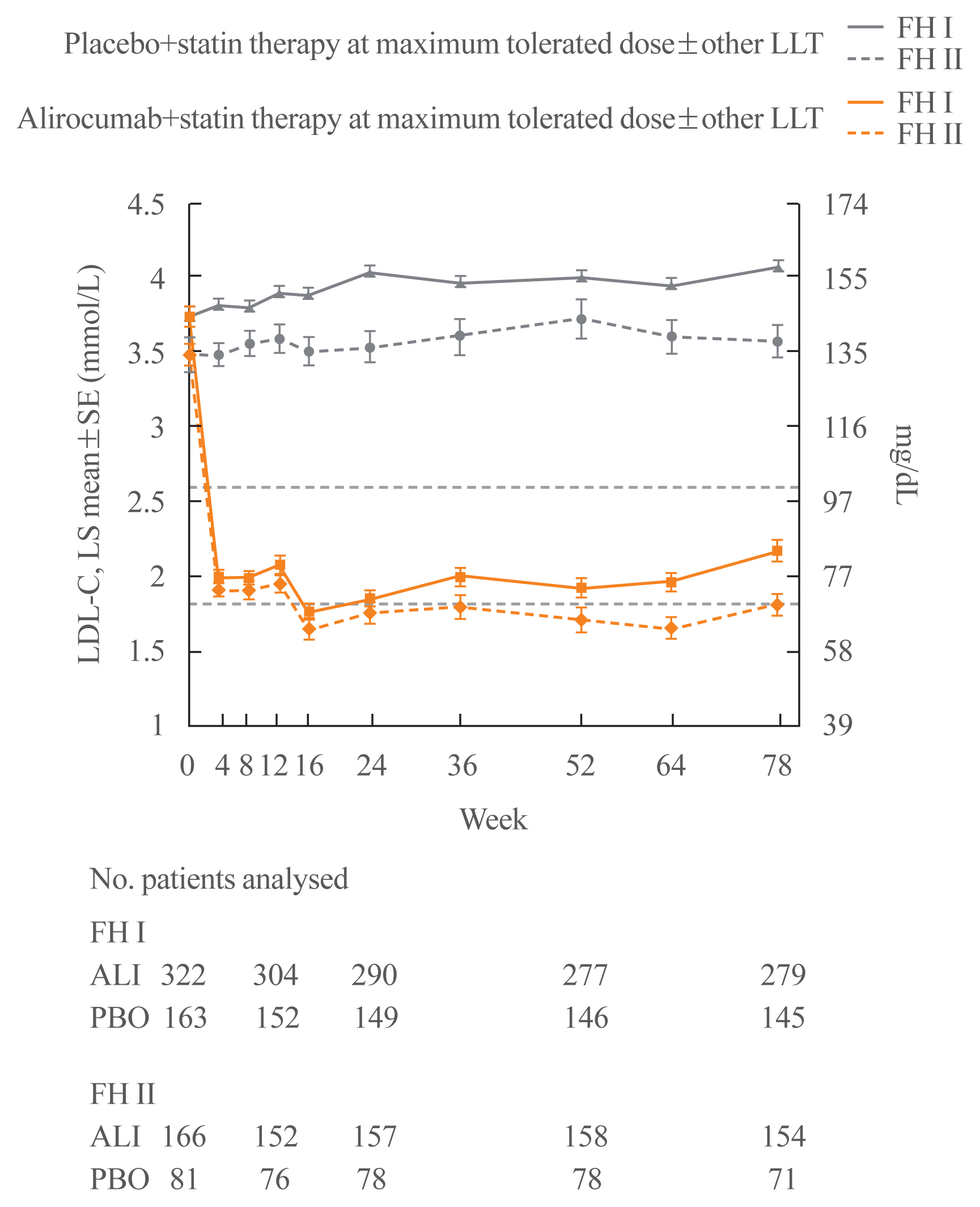
- The ODYSSEY FH I and FH II trials were phase 3 trials in 486 and 249 patients with HeFH, respectively, who were not at the appropriate LDL-C target for primary or secondary prevention despite stable high-dose statin therapy with or without other lipid-lowering therapy [72]. As in the other ODYSSEY studies, the initial alirocumab dose was 75 mg Q2W and the dose was increased to 150 mg Q2W at week 12 if the LDL-C level was ≥1.8 mmol/L (70 mg/dL) at week 8. At week 24, LDL-C levels decreased relative to placebo by 57.9% and 51.4%, respectively, in the two studies (Fig. 3). These reductions were maintained through week 78. Adverse events that resulted in discontinuation of the study drug occurred with similar frequency with alirocumab and placebo. Injection site reactions were slightly more frequent in the alirocumab groups compared to placebo in FH I (12.4% vs. 11.0%) and FH II (11.4% vs. 7.4%), but these differences were not statistically significant.
- The proportion of subjects with LDL-C <1.8 mmol/L at week 8 was 56.6% in FH I and 61.4% in FH II and the additional percentage reduction in LDL-C in subjects who had a dose increase was 15.1% in FH I and 16.9% in FH II. At week 24, an LDL-C level of <1.8 mmol/L was achieved with alirocumab by 59.8% of patients in FH I and 68.2% of patients in FH II [72].
- In the ODYSSEY HIGH FH study, patients with HeFH and LDL-C ≥160 mg/dL despite the maximally tolerated statin dose with or without other lipid-lowering therapy were randomized to alirocumab 150 mg or placebo Q2W [73]. LDL-C was reduced by 39.1% compared to placebo at week 24 and 41% of patients treated with alirocumab were able to achieve predefined LDL-C goals, which were <70 mg/dL for very-high-risk patients and <100 mg/dL for high-risk patients. The mean±SD baseline LDL-C was 196.3±57.9 mg/dL in the alirocumab group and this was reduced to a mean±standard error (SE) level of 107.0±6.7 mg/dL at 24 weeks and the reductions were maintained to week 78. Injection-site reactions were more frequent in the alirocumab group (8.3%) than in the placebo group (5.7%). These were mostly mild in severity and did not result in study medication discontinuation.
- Some patients with HeFH were also included in the ODYSSEY LONG-TERM and ODYSSEY JAPAN phase 3 studies [74,75]. There were 27/144 and 14/72 patients with HeFH treated with alirocumab (75 mg Q2W) or with placebo, respectively, in the ODYSSEY JAPAN study [75]. The mean±SE reduction in LDL-C relative to placebo in the overall study was 64.1%±2.2% at 24 weeks, and this reduction was maintained to week 52. The results for subjects with HeFH were not reported separately.
- In the ODYSSEY LONG-TERM phase 3 study, 17.7% of patients had HeFH and they showed a similar response to alirocumab 150 mg Q2W as the overall trial patients, with a mean±SE reduction of 61.9%±1.3% in LDL-C at 24 weeks compared to placebo, and the effect remained consistent over a period of 78 weeks [74].
- Of the patients with HeFH who completed the ODYSSEY LONG TERM study, 214 subsequently enrolled in the open-label extension (OLE) study, ODYSSEY OLE [76]. Following an 8-week washout period, patients were started on alirocumab 75 mg Q2W and the dose was increased to 150 mg Q2W from week 12 based on the physician’s clinical judgment. The mean LDL-C reduction from the OLE baseline at week 96 was 46.8% for patients without an increase in the alirocumab dose and 55.4% for those with an increased dose. Consistent LDL-C reductions were maintained over a treatment period of up to 4 years including the 1.5 years of the original ODYSSEY LONG TERM trial.
- In a post hoc analysis to examine whether age modified the efficacy and safety of alirocumab in patients with HeFH, pooled data from four phase 3 trials (ODYSSEY FH I, FH II, LONG TERM, and HIGH FH) were compared for the age subgroups of 18 to <45, 45 to <55, 55 to <65, and ≥65 years [77]. The mean LDL-C reductions were similar across age groups for the same alirocumab dose regimens, and treatment-emergent adverse events were similar in frequency in alirocumab- and placebo-treated patients in all age groups, with the exception that injection-site reactions were more common with alirocumab than with placebo, but declined in frequency with age.
- In a multinational study that identified 164 HeFH patients with GOF mutations in PCSK9, there was a wide range of LDL-C levels and a high risk of premature ASCVD [78]. In 13 of these patients, a single dose of alirocumab 150 mg reduced LDL-C by a mean±SE of 53.7%±11.5% compared with placebo-treated patients at 2 weeks, and after all subjects received alirocumab 150 mg Q2W for 8 weeks, LDL-C was reduced by 73.3%±16.1% from baseline (P<0.0001).
- The ODYSSEY KIDS study was a phase 2 dose-finding study in 42 pediatric HeFH patients aged 8 to 17 years with LDL-C ≥130 mg/dL (3.37 mmol/L) despite optimal statin treatment with or without other lipid-modifying therapies [79]. Alirocumab was given in various doses of 30 or 50 mg Q2W, 40 or 75 mg Q2W, 75 or 150 mg Q4W, and 150 or 300 mg Q4W chosen according to whether patients’ body weight was <50 or ≥50 kg in four cohorts. The greatest reductions in LDL-C were mean±SE 46.1%±8.3% in the group receiving 40 or 75 mg Q2W and 44.5%±7.6% in the group receiving 150 or 300 mg Q4W.
- Alirocumab 150 mg Q2W was tested in 69 adults with HoFH in the ODYSSEY HoFH trial [80]. Most patients were on high-intensity statin and ezetimibe, 10 patients were on lomitapide, and 10 patients were undergoing apheresis. The patients were randomized to alirocumab 150 mg or placebo Q2W for 12 weeks, and then all patients continued on open label alirocumab 150 mg Q2W for another 12 weeks. The mean baseline LDL-C level was 295.0 mg/dL in the alirocumab group and 259.6 mg/dL in the placebo group. The least squares mean difference in LDL-C from baseline after 12 weeks was −26.9% with alirocumab compared to 8.6% with placebo, resulting in a mean±SE reduction with alirocumab relative to placebo of 35.6%±7.8%. The mean reduction in LDL-C from baseline to week 24, when all patients had received alirocumab was 27.3%, corresponding to an absolute reduction of 67.9 mg/dL. There was wide variation in LDL-C responses.
- In a pooled analysis of eight ODYSSEY phase 3 clinical program trials in high cardiovascular risk populations with or without HeFH, alirocumab 150 mg Q2W reduced LDL-C by 60.4% compared with an increase of 0.5% with placebo [81]. There were also consistent reductions in apoB, non-high-density lipoprotein cholesterol, and Lp(a). Similar findings have been reported in other studies of alirocumab. In a pooled analysis of three double-blind phase 2 trials with alirocumab 150 mg Q2W, there was a 30% reduction in Lp(a) compared to placebo (P<0.0001) [82].
- The Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome with Alirocumab (ODYSSEY Outcomes) trial randomized patients to placebo or to alirocumab at 1 to 12 months after hospitalization for acute coronary syndrome [83]. The 15% reduction in the primary composite CV endpoint was significant (P<0.001) and all-cause mortality was also reduced after a median follow-up of 2.8 years. Patients with baseline LDL-C >100 mg/dL (2.6 mmol/L) had a greater absolute benefit with alirocumab than those with lower values for the primary composite endpoint.
PCSK9 MONOCLONAL ANTIBODIES
- Inclisiran is a long-acting small interfering RNA (siRNA) agent that prevents translation of PCSK9 messenger RNA (mRNA), leading to decreased PCSK9 protein concentrations in hepatocytes and plasma. It was previously known as ALN-PCSsc, which was developed from ALN-PCS. It consists of two synthetic RNA strands conjugated to triantennary N-acetylgalactosamine (GalNAc), which is a ligand for the asialoglycoprotein receptor (ASGPR) expressed on hepatocytes, providing high-affinity binding with rapid and selective uptake of the compound into the liver. In the liver, it forms the RNA-inducing silencing complex (RISC), which prevents intracellular translation of PCSK9 mRNA to protein (Fig. 1) [84].
- Inclisiran is undergoing development in the ORION clinical development program [85]. A phase 1 dose-finding study showed that SC doses of 300 mg or more can reduce LDL-C by more than 50% for at least 6 months [86]. The ORION-1 trial was a phase 2 study that examined the effects of different doses of inclisiran in subjects with high cardiovascular risk and elevated LDL-C despite maximally tolerated statin therapy, including 6% of subjects with FH [87]. Inclisiran produced dose-dependent reductions in plasma PCSK9 and LDL-C levels, and the 2-dose 300 mg regimen gave the greatest reduction in LDL-C. Injection-site reactions occurred in 5% of the subjects given inclisiran. A 1-year follow-up study of the ORION-1 trial showed that inclisiran produced durable reductions in LDL-C over 1 year [88]. The effects of inclisiran in patients with FH are shown in Table 3.
- In the phase 2 ORION-2 Pilot study in four patients with HoFH and LDL-C >500 mg/dL, inclisiran 300 mg was given on day 1 and day 90 or 104 and the change in LDL-C at day 180 varied between patients from −37.0% to 3.3% [89]. The ORION-5 study (NCT03851705) is an ongoing phase 3 trial in 56 patients with HoFH who are being treated with inclisiran 300 mg or placebo on days 1 and 90 in Part 1, while in Part 2 all the study subjects will receive a dose of 300 mg inclisiran SC on day 270, day 450, and day 630 [90].
- In ORION-9, 482 patients with HeFH received placebo or inclisiran 300 mg SC at baseline, 3 months later, and then every 6 months for a total of four doses [91]. At day 510, the mean reduction in LDL-C with inclisiran was 39.7% compared with an increase of 8.2% in the placebo group, giving a mean placebo-adjusted LDL-C reduction of 47.9% at day 510 and a time-averaged LDL-C reduction of 44.3% over the 18-month trial. In a single patient with a PCSK9 GOF variant, the placebo-adjusted LDL-C reduction was 89.7%.
- The ORION-4 trial (NCT03705234) is an ongoing cardiovascular outcome trial to examine the effect of inclisiran on cardiovascular events in about 15,000 patients at very high cardiovascular risk over 5 years [92].
- Overall, inclisiran and the mAbs that inhibit PCSK9 are very well-tolerated apart from a small number of injection-site reactions, which are generally mild. These treatments reduce LDL-C by more than 50% in HeFH patients when given in addition to treatment with statins and other lipid-lowering drugs and have a similar effect to that seen in subjects without FH. The reductions in LDL-C in HoFH patients are smaller and appear to depend on the type of mutation in the LDLR gene. PCSK9 inhibitors therefore represent a very useful therapy for the majority of patients with FH. Mendelian randomization studies suggested that genetic variants in PCSK9 associated with lower LDL-C levels were also associated with an increased risk of new-onset type 2 diabetes [93,94]. This has not been seen in clinical studies with the PCSK9 inhibitors, but is known to be a risk with statin therapy [95,96].
INCLISIRAN
- The PCSK9 inhibitors can bring a substantial number of patients with HeFH to goal in combination with high-intensity statin therapy and ezetimibe. However, very few patients with HoFH will reach the LDL-C target with the addition of PCSK9 inhibitors and other treatments are usually required. Currently, lomitapide and apheresis are the main options. Lomitapide is a microsomal triglyceride transfer protein inhibitor given orally [97]. It prevents the formation of apoB-containing lipoproteins in the intestines and liver. There are concerns over adverse effects, which were mainly gastrointestinal problems and hepatic steatosis, but recent reports show that in clincal practice it has been used in lower doses than in the phase 3 study, with reductions in LDL-C of more than 50% in many patients and less severe adverse events [98]. Data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER) show that the median dose was 10 mg and the mean reduction in LDL-C was 33% overall, or 45% in patients remaining on lomitapide treatment [99].
- Bempedoic acid, previously known as ETC-1002, is an inhibitor of ATP citrate lyase (ACL), which is an enzyme in the cholesterol synthesis pathway upstream of the statin target, 3-hydroxy-3-methylglutaryl-CoA reductase [100]. It was approved by the FDA for the treatment of adults with HeFH or established ASCVD who require additional lowering of LDL-C in February 2020 [101]. It is given in the form of an inactive prodrug that is activated selectively in the liver, so it is less likely to cause the skeletal muscle problems that may occur with statins [102].
- Bempedoic acid produces additional reductions in LDL-C of about 18% to 20% when added to statins and has an additive effect when given with ezetimibe [103]. It may prove to be particularly useful in FH patients with statin intolerance. A cardiovascular outcome study with bempedoic acid, Cholesterol Lowering via BEmpedoic Acid, an ACL-inhibiting Regimen (CLEAR) Outcomes (NCT02993406), is ongoing [104].
- Other promising treatments, which were origally developed to reduce triglycerides but may be particularly relavant for patients with HoFH, are those that target angiopoietin-like protein 3 (ANGPTL3), which inhibits hepatic and endothelial lipases. Evinacumab is a mAb directed against ANGPTL3. In a study in 65 patients with HoFH receiving stable lipid-lowering therapy, intravenous evinacumab 15 mg/kg Q4W for 24 weeks reduced LDL-C relative to placebo by 49.0% (95% CI, 33.1% to 65.0%; P<0.001) with no major adverse effects [105].
- Vupanorsen, or AKCEA-ANGPTL3-LRx, is a GalNAc-modified antisense oligonucleotide (ASO) that is more potent than an earlier non-GalNAc-modified ANGPTL3 ASO. An early study showed it reduced triglycerides in healthy subjects with and without elevated triglyceride levels when given SC once weekly [106]. Vupanorsen at a dose of 20 mg QW or higher doses Q4W was effective in reducing triglycerides and other atherogenic lipoproteins in patients with diabetes and hypertriglyceridemia without any major adverse effects [107]. The reduction in LDL-C was only about 7%, and HDL-C was also reduced.
- ARO-ANG3 is a siRNA with a ligand for hepatic ASGPRs that is in development. Preliminary results showed that it reduced LDL-C by 23% to 37% in 17 patients with HeFH and was well tolerated [108]. Like inclisiran, ARO-ANG3 is likely to have a longer duration of action than the ASOs and may therefore prove to be a useful addition in patients with HeFH and HoFH.
ADDITIONAL TREATMENTS
- Patients with FH are at high or very high risk for ASCVD. Many patients with HeFH do not reach LDL-C targets with statins and ezetimibe, and most patients with HoFH do not reach LDL-C targets with currently available treatments including apheresis. Inhibition of PCSK9 with mAbs or siRNA can reduce LDL-C in patients with HeFH to a similar extent as in those without FH, by about 50% to 60%, and the effect has been durable over the period of the studies. The reduction in LDL-C is less in patients with HoFH (20% to 35% on average) and varies according to the type of mutation. PCSK9 inhibitors also reduce Lp(a), which may be an advantage in many patients with FH, and they have a good safety profile apart from a relatively low rate of mild injection-site reactions. Other treatments that can reduce LDL-C without involving the LDLR are needed for patients with HoFH, and ANGPTL3 inhibitors may prove to have an important role.
CONCLUSIONS
-
Acknowledgements
- This work was funded by faculty research funding from the Faculty of Medicine, Macau University of Science and Technology.
-
CONFLICTS OF INTEREST
Brian Tomlinson has acted as consultant or speaker for Amgen Inc., Kowa, and Merck Serono, for which he received honoraria. The other authors have no conflicts to disclose.
Article information

| Study, duration, and reference | Patients, number and type | Treatment | Change in LDL-C, %a |
|---|---|---|---|
| RUTHERFORD, 12 weeks, Raal et al. (2012) [56] | 56 HeFH | Placebo Q4W | 1.1±2.9 |
| 55 HeFH | E 350 mg Q4W | −42.7±2.9 | |
| 56 HeFH | E 420 mg Q4W | −55.2±2.9 | |
|
|
|||
| RUTHERFORD-2, 12 weeks, Raal et al. (2015) [58] | 54 HeFH | Placebo Q2W | −1.1 (−5.8 to 3.7) |
| 110 HeFH | E 140 mg Q2W | −61.2 (−64.6 to −57.9) | |
| 55 HeFH | Placebo QM | 2.3 (−2.5 to 7.1) | |
| 110 HeFH | E 420 mg QM | −63.3 (−66.6 to −59.9) | |
|
|
|||
| HAUSER-RCT, 24 weeks, Santos et al. (2020) [60] | 53 HeFHb | Placebo QM | −5.9 (−11.1 to −0.6) |
| 104 HeFHb | E 420 mg QM | −48.0 (−51.7 to −44.2) | |
|
|
|||
| TESLA, 12 weeks, Stein et al. (2013) [61] | 8 HoFH | E 420 mg Q4W | −16.5±19.0c |
| 8 HoFH | E 420 mg Q2W | −13.9±27.2c | |
|
|
|||
| TESLA Part B, 12 weeks, Raal et al. (2015) [62] | 16 HoFH | Placebo QM | 7.9 (−2.7 to 18.5) |
| 33 HoFH | E 420 mg Q4W | −23.1 (30.7 to −15.4) | |
|
|
|||
| TAUSSIG, 12 weeks, Raal et al. (2017) [63] | 106 HoFH | E 420 mg Q4W | −20.6±24.4c |
LDL-C, low-density lipoprotein cholesterol; RUTHERFORD, Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder; HeFH, heterozygous familial hypercholesterolemia; Q4W, every 4 weeks; E, evolocumab; Q2W, every 2 weeks; QM, every month; TESLA, The Trial Evaluating PCSK9 Antibody in Subjects With LDL Receptor Abnormalities; HoFH, homozygous familial hypercholesterolemia; TAUSSIG, Trial Assessing Long-Term Use of PCSK9 Inhibition in Subjects with Genetic LDL Disorders.
a Values are expressed as least-squares mean±standard error or (95% CI) unless indicated;
b Pediatric heterozygous familial hypercholesterolemia;
c Values are expressed as mean±standard deviation changes.
| Study, duration, and reference | Patients, number and type | Treatment | Change in LDL-C, %a |
|---|---|---|---|
| Phase 2 study, 12 weeks, Stein et al. (2012) [70] | 15 HeFH | Placebo Q2W | −10.65±5.04 |
| 15 HeFH | A 150 mg Q4W | −28.87±5.08 | |
| 16 HeFH | A 200 mg Q4W | −31.54±4.91 | |
| 15 HeFH | A 300 mg Q4W | −42.53±5.09 | |
| 16 HeFH | A 150 mg Q2W | −67.90±4.85 | |
|
|
|||
| ODYSSEY FH I, 24 weeks; ODYSSEY FH II, 24 weeks, Kastelein et al. (2015) [72] | 163 HeFH | Placebo Q2W | 9.1±2.2 |
| 322 HeFH | A 75/150 mg Q2W | −48.8±1.6 | |
| 55 HeFH | Placebo Q2W | 2.8±2.8 | |
| 166 HeFH | A 75/150 mg Q2W | −48.7±1.9 | |
|
|
|||
| ODYSSEY HIGH FH, 24 weeks, Ginsberg et al. (2016) [73] | 35 HeFH | Placebo Q2W | −6.6±4.9 |
| 71 HeFH | A 150 mg Q2W | −45.7±3.5 | |
|
|
|||
| ODYSSEY KIDS, 8 weeks, Daniels et al. (2020) [79] | 10 HeFHb | A 30/50 mg Q2W | −21.2±7.9 |
| 10 HeFHb | A 40/75 mg Q2W | −46.1±8.3 | |
| 11 HeFHb | A 75/150 mg Q4W | −7.8±7.6 | |
| 11 HeFHb | A 150/300 mg Q4W | −44.5±7.6 | |
|
|
|||
| ODYSSEY HoFH, 12 weeks, Blom et al. (2020) [80] | 24 HoFH | Placebo Q2W | 8.9±19.0 |
| 45 HoFH | A 150 mg Q2W | −26.9±4.6 | |
LDL-C, low-density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolemia; Q2W, every 2 weeks; A, alirocumab; Q4W, every 4 weeks; ODYSSEY FH I, Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in Patients With Heterozygous Familial Hypercholesterolemia Not Adequately Controlled With Their Lipid-Modifying Therapy; ODYSSEY FH II, Study of Alirocumab (REGN727/SAR236553) in Patients With heFH (Heterozygous Familial Hypercholesterolemia) Who Are Not Adequately Controlled With Their LMT (Lipid-Modifying Therapy); ODYSSEY HIGH FH, Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in Patients With Heterozygous Familial Hypercholesterolemia; ODYSSEY KIDS, An 8-Week Dose-Finding Study to Evaluate the Efficacy and Safety of Alirocumab in Children and Adolescents With Heterozygous Familial Hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia.
a Values are expressed as least-squares mean±standard error;
b Pediatric heterozygous familial hypercholesterolemia.
| Study, duration, and reference | Patients, number and type | Treatment | Change in LDL-C, % |
|---|---|---|---|
| ORION-2, 180 days, Hovingh et al. (2020) [89] | 4 HoFH | I 300 mg on days 1 and 90/104 | −37.0 to 3.3 |
|
|
|||
| ORION-5, 24 months, (NCT03851705) [90] | 56 HoFH randomized 2:1 to I or placebo | Placebo on days 1, 90 | Awaited |
| I 300 mg on days 1, 90, 270, 450, 630 | |||
|
|
|||
| ORION-9, 18 months, Raal et al. (2020) [73] | 240 HeFH | Placebo on days 1, 90, 270, 450 | 8.2 (4.3 to 12.2) |
| 242 HeFH | I 300 mg on days 1, 90, 270, 450 | −39.7 (−43.7 to −35.7) | |
- 1. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–3490a.PubMedPMC
- 2. Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 2017;7:e016461.ArticlePubMedPMC
- 3. Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, et al. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol 2018;72:662–80.PubMed
- 4. Foody JM, Vishwanath R. Familial hypercholesterolemia/autosomal dominant hypercholesterolemia: molecular defects, the LDL-C continuum, and gradients of phenotypic severity. J Clin Lipidol 2016;10:970–86.ArticlePubMed
- 5. Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management: a position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014;35:2146–57.ArticlePubMedPMC
- 6. Sjouke B, Kusters DM, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J 2015;36:560–5.ArticlePubMed
- 7. Lee SH. Update on familial hypercholesterolemia: diagnosis, cardiovascular risk, and novel therapeutics. Endocrinol Metab (Seoul) 2017;32:36–40.ArticlePubMedPMC
- 8. Tomlinson B, Chan JC, Chan WB, Chen WW, Chow FC, Li SK, et al. Guidance on the management of familial hypercholesterolaemia in Hong Kong: an expert panel consensus viewpoin. Hong Kong Med J 2018;24:408–15.PubMed
- 9. Rhee EJ, Kim HC, Kim JH, Lee EY, Kim BJ, Kim EM, et al. 2018 Guidelines for the management of dyslipidemia. Korean J Intern Med 2019;34:723–71.ArticlePubMedPMC
- 10. Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation 2015;132:2167–92.ArticlePubMed
- 11. Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017. J Atheroscler Thromb 2018;25:751–70.ArticlePubMedPMC
- 12. Nordestgaard BG, Benn M. Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J 2017;38:1580–3.ArticlePubMed
- 13. Iacocca MA, Chora JR, Carrie A, Freiberger T, Leigh SE, Defesche JC, et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat 2018;39:1631–40.ArticlePubMedPMC
- 14. Huang CC, Charng MJ. Genetic diagnosis of familial hypercholesterolemia in Asia. Front Genet 2020;11:833.ArticlePubMedPMC
- 15. Tomlinson B, Hu M, Chow E. Current status of familial hypercholesterolemia in Chinese populations. Curr Opin Lipidol 2019;30:94–100.ArticlePubMed
- 16. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004;160:407–20.ArticlePubMed
- 17. Soutar AK, Naoumova RP, Traub LM. Genetics, clinical phenotype, and molecular cell biology of autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol 2003;23:1963–70.ArticlePubMed
- 18. Stitziel NO, Fouchier SW, Sjouke B, Peloso GM, Moscoso AM, Auer PL, et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol 2013;33:2909–14.ArticlePubMedPMC
- 19. Strebinger G, Muller E, Feldman A, Aigner E. Lysosomal acid lipase deficiency: early diagnosis is the key. Hepat Med 2019;11:79–88.PubMedPMC
- 20. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003;34:154–6.ArticlePubMed
- 21. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003;100:928–33.ArticlePubMedPMC
- 22. Pijlman AH, Huijgen R, Verhagen SN, Imholz BP, Liem AH, Kastelein JJ, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in the Netherlands. Atherosclerosis 2010;209:189–94.ArticlePubMed
- 23. Wang J, Dron JS, Ban MR, Robinson JF, McIntyre AD, Alazzam M, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol 2016;36:2439–45.ArticlePubMed
- 24. Trinder M, Paquette M, Cermakova L, Ban MR, Hegele RA, Baass A, et al. Polygenic contribution to low-density lipoprotein cholesterol levels and cardiovascular risk in monogenic familial hypercholesterolemia. Circ Genom Precis Med 2020;13:515–23.ArticlePubMedPMC
- 25. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020;41:111–88.ArticlePubMedPDF
- 26. Perez de Isla L, Alonso R, Mata N, Fernandez-Perez C, Muniz O, Diaz-Diaz JL, et al. Predicting cardiovascular events in familial hypercholesterolemia: the SAFEHEART registry (Spanish familial hypercholesterolemia cohort study). Circulation 2017;135:2133–44.PubMed
- 27. Alonso R, Andres E, Mata N, Fuentes-Jimenez F, Badimon L, Lopez-Miranda J, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol 2014;63:1982–9.PubMed
- 28. Yeang C, Willeit P, Tsimikas S. The interconnection between lipoprotein(a), lipoprotein(a) cholesterol and true LDL-cholesterol in the diagnosis of familial hypercholesterolemia. Curr Opin Lipidol 2020;31:305–12.ArticlePubMed
- 29. Tsimikas S, Gordts PL, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J 2020;41:2275–84.ArticlePubMed
- 30. Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008;337:a2423.ArticlePubMedPMC
- 31. Luirink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, de Groot E, et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med 2019;381:1547–56.ArticlePubMed
- 32. Oh J, Lee CJ, Kim DI, Rhee MY, Lee BK, Ahn Y, et al. Target achievement with maximal statin-based lipid-lowering therapy in Korean patients with familial hypercholesterolemia: a study supported by the Korean Society of Lipid and Atherosclerosis. Clin Cardiol 2017;40:1291–6.ArticlePubMedPMC
- 33. Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation 2011;124:2202–7.ArticlePubMed
- 34. Ito MK, Santos RD. PCSK9 inhibition with monoclonal antibodies: modern management of hypercholesterolemia. J Clin Pharmacol 2017;57:7–32.ArticlePubMed
- 35. Landmesser U, Chapman MJ, Farnier M, Gencer B, Gielen S, Hovingh GK, et al. European Society of Cardiology/European Atherosclerosis Society task force consensus statement on proprotein convertase subtilisin/kexin type 9 inhibitors: practical guidance for use in patients at very high cardiovascular risk. Eur Heart J 2017;38:2245–55.ArticlePubMed
- 36. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005;37:161–5.ArticlePubMedPDF
- 37. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–72.ArticlePubMed
- 38. Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol 2010;55:2833–42.PubMed
- 39. Abifadel M, Rabes JP, Devillers M, Munnich A, Erlich D, Junien C, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 2009;30:520–9.ArticlePubMed
- 40. Lambert G, Charlton F, Rye KA, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis 2009;203:1–7.ArticlePubMed
- 41. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008;283:2363–72.ArticlePubMed
- 42. Qian YW, Schmidt RJ, Zhang Y, Chu S, Lin A, Wang H, et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res 2007;48:1488–98.ArticlePubMed
- 43. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2004;24:1454–9.ArticlePubMed
- 44. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res 2008;49:399–409.ArticlePubMed
- 45. Seidah NG, Awan Z, Chretien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res 2014;114:1022–36.ArticlePubMed
- 46. Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A 2005;102:5374–9.ArticlePubMedPMC
- 47. Raal F, Panz V, Immelman A, Pilcher G. Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc 2013;2:e000028.ArticlePubMedPMC
- 48. Davignon J, Dubuc G. Statins and ezetimibe modulate plasma proprotein convertase subtilisin kexin-9 (PCSK9) levels. Trans Am Clin Climatol Assoc 2009;120:163–73.PubMedPMC
- 49. Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies: what cardiologists need to know. Circulation 2013;127:2222–30.ArticlePubMed
- 50. Ridker PM, Revkin J, Amarenco P, Brunell R, Curto M, Civeira F, et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N Engl J Med 2017;376:1527–39.ArticlePubMed
- 51. Ridker PM, Tardif JC, Amarenco P, Duggan W, Glynn RJ, Jukema JW, et al. Lipid-reduction variability and antidrug-antibody formation with bococizumab. N Engl J Med 2017;376:1517–26.ArticlePubMed
- 52. Fazio S, Robertson DG, Joh T, Wan H, Riel T, Forgues P, et al. Effects of 12 weeks of treatment with intravenously administered bococizumab, a humanized monoclonal antibody blocking proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects on high-dose statin. Cardiovasc Ther 2018;36:e12308.Article
- 53. Ridker PM, Rose LM, Kastelein JJP, Santos RD, Wei C, Revkin J, et al. Cardiovascular event reduction with PCSK9 inhibition among 1578 patients with familial hypercholesterolemia: results from the SPIRE randomized trials of bococizumab. J Clin Lipidol 2018;12:958–65.ArticlePubMed
- 54. Stein EA, Wasserman SM, Dias C, Scott R, Raal F. AMG-145, Evolocumab. Drugs Future 2013;38:451–9.
- 55. Tomlinson B, Hu M, Zhang Y, Chan P, Liu ZM. Evolocumab for the treatment of hypercholesterolemia. Expert Opin Biol Ther 2017;17:1447–61.ArticlePubMed
- 56. Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012;126:2408–17.ArticlePubMed
- 57. Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet 2012;380:1995–2006.ArticlePubMed
- 58. Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:331–40.ArticlePubMed
- 59. Lee T, Iacocca MA, Ban MR, Hegele RA. Efficacy of evolocumab in monogenic vs polygenic hypercholesterolemia. CJC Open 2019;1:115–8.ArticlePubMedPMC
- 60. Santos RD, Ruzza A, Hovingh GK, Wiegman A, Mach F, Kurtz CE, et al. Evolocumab in pediatric heterozygous familial hypercholesterolemia. N Engl J Med 2020;383:1317–27.ArticlePubMed
- 61. Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation 2013;128:2113–20.ArticlePubMed
- 62. Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:341–50.ArticlePubMed
- 63. Raal FJ, Hovingh GK, Blom D, Santos RD, Harada-Shiba M, Bruckert E, et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol 2017;5:280–90.ArticlePubMed
- 64. Santos RD, Stein EA, Hovingh GK, Blom DJ, Soran H, Watts GF, et al. Long-term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol 2020;75:565–74.ArticlePubMed
- 65. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 2017;376:1713–22.ArticlePubMed
- 66. Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Seidah NG, et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. J Lipid Res 2016;57:1086–96.ArticlePubMedPMC
- 67. Tomlinson B, Hu M, Zhang Y, Chan P, Liu ZM. Alirocumab for the treatment of hypercholesterolemia. Expert Opin Biol Ther 2017;17:633–43.ArticlePubMed
- 68. Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med 2012;366:1108–18.ArticlePubMed
- 69. Rey J, Poitiers F, Paehler T, Brunet A, DiCioccio AT, Cannon CP, et al. Relationship between low-density lipoprotein cholesterol, free proprotein convertase subtilisin/kexin type 9, and alirocumab levels after different lipid-lowering strategies. J Am Heart Assoc 2016;5:e003323.ArticlePubMedPMC
- 70. Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet 2012;380:29–36.ArticlePubMed
- 71. Dufour R, Bergeron J, Gaudet D, Weiss R, Hovingh GK, Qing Z, et al. Open-label therapy with alirocumab in patients with heterozygous familial hypercholesterolemia: results from three years of treatment. Int J Cardiol 2017;228:754–60.ArticlePubMed
- 72. Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015;36:2996–3003.ArticlePubMedPMC
- 73. Ginsberg HN, Rader DJ, Raal FJ, Guyton JR, Baccara-Dinet MT, Lorenzato C, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia and LDL-C of 160 mg/dl or higher. Cardiovasc Drugs Ther 2016;30:473–83.ArticlePubMedPMC
- 74. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015;372:1489–99.ArticlePubMed
- 75. Teramoto T, Kobayashi M, Tasaki H, Yagyu H, Higashikata T, Takagi Y, et al. Efficacy and safety of alirocumab in Japanese patients with heterozygous familial hypercholesterolemia or at high cardiovascular risk with hypercholesterolemia not adequately controlled with statins: ODYSSEY JAPAN randomized controlled trial. Circ J 2016;80:1980–7.ArticlePubMed
- 76. Dufour R, Hovingh GK, Guyton JR, Langslet G, Baccara-Dinet MT, Din-Bell C, et al. Individualized low-density lipoprotein cholesterol reduction with alirocumab titration strategy in heterozygous familial hypercholesterolemia: results from an open-label extension of the ODYSSEY LONG TERM trial. J Clin Lipidol 2019;13:138–47.ArticlePubMed
- 77. Ginsberg HN, Tuomilehto J, Hovingh GK, Cariou B, Santos RD, Brown AS, et al. Impact of age on the efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia. Cardiovasc Drugs Ther 2019;33:69–76.ArticlePubMedPMC
- 78. Hopkins PN, Defesche J, Fouchier SW, Bruckert E, Luc G, Cariou B, et al. Characterization of autosomal dominant hypercholesterolemia caused by PCSK9 gain of function mutations and its specific treatment with alirocumab, a PCSK9 monoclonal antibody. Circ Cardiovasc Genet 2015;8:823–31.ArticlePubMedPMC
- 79. Daniels S, Caprio S, Chaudhari U, Manvelian G, Baccara-Dinet MT, Brunet A, et al. PCSK9 inhibition with alirocumab in pediatric patients with heterozygous familial hypercholesterolemia: the ODYSSEY KIDS study. J Clin Lipidol 2020;14:322–30.ArticlePubMed
- 80. Blom DJ, Harada-Shiba M, Rubba P, Gaudet D, Kastelein JJ, Charng MJ, et al. Efficacy and safety of alirocumab in adults with homozygous familial hypercholesterolemia: the ODYSSEY HoFH trial. J Am Coll Cardiol 2020;76:131–42.PubMed
- 81. Farnier M, Gaudet D, Valcheva V, Minini P, Miller K, Cariou B. Efficacy of alirocumab in high cardiovascular risk populations with or without heterozygous familial hypercholesterolemia: pooled analysis of eight ODYSSEY Phase 3 clinical program trials. Int J Cardiol 2016;223:750–7.ArticlePubMed
- 82. Gaudet D, Kereiakes DJ, McKenney JM, Roth EM, Hanotin C, Gipe D, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol 2014;114:711–5.ArticlePubMed
- 83. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med 2018;379:2097–107.ArticlePubMed
- 84. Nishikido T, Ray KK. Inclisiran for the treatment of dyslipidemia. Expert Opin Investig Drugs 2018;27:287–94.ArticlePubMed
- 85. German CA, Shapiro MD. Small interfering RNA therapeutic inclisiran: a new approach to targeting PCSK9. BioDrugs 2020;34:1–9.ArticlePubMed
- 86. Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med 2017;376:41–51.ArticlePubMed
- 87. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med 2017;376:1430–40.ArticlePubMed
- 88. Ray KK, Stoekenbroek RM, Kallend D, Nishikido T, Leiter LA, Landmesser U, et al. Effect of 1 or 2 doses of inclisiran on low-density lipoprotein cholesterol levels: one-year follow-up of the ORION-1 randomized clinical trial. JAMA Cardiol 2019;4:1067–75.ArticlePubMedPMC
- 89. Hovingh GK, Lepor NE, Kallend D, Stoekenbroek RM, Wijngaard PL, Raal FJ. Inclisiran durably lowers low-density lipoprotein cholesterol and proprotein convertase subtilisin/kexin type 9 expression in homozygous familial hypercholesterolemia: the ORION-2 pilot study. Circulation 2020;141:1829–31.ArticlePubMed
- 90. Clinicaltrials.gov. A study of inclisiran in participants with homozygous familial hypercholesterolemia (HoFH) (ORION-5) [Internet] Bethesda: National Library of Medicine; 2021 [cited 2021 Mar 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03851705.
- 91. Raal FJ, Kallend D, Ray KK, Turner T, Koenig W, Wright RS, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med 2020;382:1520–30.ArticlePubMed
- 92. Clinicaltrials.gov. A randomized trial assessing the effects of inclisiran on clinical outcomes among people with cardiovascular disease (ORION-4) [Internet] Bethesda: National Library of Medicine; 2021 [cited 2021 Mar 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03705234.
- 93. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016;375:2144–53.ArticlePubMed
- 94. Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol 2017;5:97–105.PubMedPMC
- 95. Sattar N, Toth PP, Blom DJ, Koren MJ, Soran H, Uhart M, et al. Effect of the proprotein convertase subtilisin/kexin type 9 inhibitor evolocumab on glycemia, body weight, and new-onset diabetes mellitus. Am J Cardiol 2017;120:1521–7.ArticlePubMed
- 96. Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol 2011;22:460–6.PubMed
- 97. Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014;129:1022–32.ArticlePubMed
- 98. Stefanutti C. Lomitapide: a microsomal triglyceride transfer protein inhibitor for homozygous familial hypercholesterolemia. Curr Atheroscler Rep 2020;22:38.ArticlePubMedPMCPDF
- 99. Underberg JA, Cannon CP, Larrey D, Makris L, Blom D, Phillips H. Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: five-year data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER). J Clin Lipidol 2020;14:807–17.ArticlePubMed
- 100. Saeed A, Ballantyne CM. Bempedoic acid (ETC-1002): a current review. Cardiol Clin 2018;36:257–64.PubMed
- 101. US Food and Drug Administration. Drugs@FDA: FDA-approved drugs [Internet] Silver Spring: FDA; 2021 [cited 2021 Mar 30]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=211616.
- 102. Pinkosky SL, Newton RS, Day EA, Ford RJ, Lhotak S, Austin RC, et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun 2016;7:13457.ArticlePubMedPMC
- 103. Thompson PD, MacDougall DE, Newton RS, Margulies JR, Hanselman JC, Orloff DG, et al. Treatment with ETC-1002 alone and in combination with ezetimibe lowers LDL cholesterol in hypercholesterolemic patients with or without statin intolerance. J Clin Lipidol 2016;10:556–67.ArticlePubMed
- 104. Clinicaltrials.gov. Evaluation of major cardiovascular events in patients with, or at high risk for, cardiovascular disease who are statin intolerant treated with bempe acid (ETC-c1002) or placebo (CLEAR Outcomes) [Internet] Bethesda: National Library of Medicine; 2020 [cited 2021 Mar 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT02993406.
- 105. Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJ, Rubba P, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med 2020;383:711–20.ArticlePubMed
- 106. Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med 2017;377:222–32.ArticlePubMed
- 107. Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, Hurh E, Kingsbury J, Bartlett VJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J 2020;41:3936–45.ArticlePubMedPMC
- 108. Watts GF, Schwabe C, Scott R, Gladding P, Sullivan D, Baker J, et al. Pharmacodynamic effect of ARO-ANG3, an investigational RNA interference targeting hepatic angiopoietin-like protein 3, in patients with hypercholesterolemia. Circulation 2020;142(Suppl 3):A15751.
References
Figure & Data
References
Citations
- Phenotypic homozygous familial hypercholesterolemia successfully treated with proprotein convertase subtilisin/kexin type 9 inhibitors
Ryosuke Tani, Keiji Matsunaga, Yuta Toda, Tomoko Inoue, Hai Ying Fu, Tetsuo Minamino
Clinical Case Reports.2024;[Epub] CrossRef - Targeting Lipoprotein(a): Can RNA Therapeutics Provide the Next Step in the Prevention of Cardiovascular Disease?
Henriette Thau, Sebastian Neuber, Maximilian Y. Emmert, Timo Z. Nazari-Shafti
Cardiology and Therapy.2024; 13(1): 39. CrossRef - Technologies of gene editing and related clinical trials for the treatment of genetic and acquired diseases: a systematic review
Wessam Sharaf-Eldin
Egyptian Journal of Medical Human Genetics.2024;[Epub] CrossRef - Qualitative and Quantitative Effects of PCSK9 Inhibitors in familial Hypercholesterolemia: a Synthetic Review
Aamina Shakir, Kyle Barron, Kalgi Modi
Current Problems in Cardiology.2023; 48(4): 101550. CrossRef - Inhibition of PCSK9 Improves the Development of Pulmonary Arterial Hypertension Via Down-Regulating Notch3 Expression
Peng Ye, Xiao-Min Jiang, Wei-Chun Qian, Juan Zhang
Cardiovascular Drugs and Therapy.2023;[Epub] CrossRef - Barriers and shortcomings in access to cardiovascular management and prevention for familial hypercholesterolemia during the COVID‐19 pandemic
Helen Huang, Keith S. K. Leung, Tulika Garg, Adele Mazzoleni, Goshen D. Miteu, Farida Zakariya, Wireko A. Awuah, Elaine T. S. Yin, Faaraea Haroon, Zarish Hussain, Narjiss Aji, Vikash Jaiswal, Gary Tse
Clinical Cardiology.2023; 46(8): 831. CrossRef - Familial Hypercholesterolemia in Children. The Current State of the Problem
Dinara I. Sadykova, Karina R. Salakhova, Liliya F. Galimova, Eugeniya S. Slastnikova, Chulpan D. Khaliullina
Current Pediatrics.2023; 22(3): 231. CrossRef - Long-term safety and effectiveness of alirocumab and evolocumab in familial hypercholesterolemia (FH) in Belgium
Marc Snel, Olivier S. Descamps
Acta Cardiologica.2023; : 1. CrossRef - PCSK9 inhibitors revisited: Effectiveness and safety of PCSK9 inhibitors in a real-life Spanish cohort
Juan Vicente-Valor, Xandra García-González, Sara Ibáñez-García, María Esther Durán-García, Ana de Lorenzo-Pinto, Carmen Rodríguez-González, Irene Méndez-Fernández, Juan Carlos Percovich-Hualpa, Ana Herranz-Alonso, María Sanjurjo-Sáez
Biomedicine & Pharmacotherapy.2022; 146: 112519. CrossRef - Development of small-molecule PCSK9 inhibitors for the treatment of hypercholesterolemia
Shakir Ahamad, Shintu Mathew, Waqas A. Khan, Kishor Mohanan
Drug Discovery Today.2022; 27(5): 1332. CrossRef - The biological relevance of PCSK9: when less is better…
Majambu Mbikay, Michel Chrétien
Biochemistry and Cell Biology.2022; 100(3): 189. CrossRef - Fenofibrate add-on to statin treatment is associated with low all-cause death and cardiovascular disease in the general population with high triglyceride levels
Kyung-Soo Kim, Sangmo Hong, Kyungdo Han, Cheol-Young Park
Metabolism.2022; 137: 155327. CrossRef - Homozygous Familial Hypercholesterolemia
Lisa Young, Emily E. Brown, Seth S. Martin
JACC: Case Reports.2022; 4(23): 101666. CrossRef - Familial Hypercholesterolemia and Its Current Diagnostics and Treatment Possibilities: A Literature Analysis
Kristina Zubielienė, Gintarė Valterytė, Neda Jonaitienė, Diana Žaliaduonytė, Vytautas Zabiela
Medicina.2022; 58(11): 1665. CrossRef - Efficacy and Safety of Alirocumab in Children and Adolescents With Homozygous Familial Hypercholesterolemia: Phase 3, Multinational Open-Label Study
Eric Bruckert, Sonia Caprio, Albert Wiegman, Min-Ji Charng, Cézar A. Zárate-Morales, Marie T. Baccara-Dinet, Garen Manvelian, Anne Ourliac, Michel Scemama, Stephen R. Daniels
Arteriosclerosis, Thrombosis, and Vascular Biology.2022; 42(12): 1447. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite