Articles
- Page Path
- HOME > Endocrinol Metab > Volume 36(5); 2021 > Article
-
Original ArticleThyroid Whole-Exome Sequencing in Papillary Microcarcinoma: Potential Early Biomarkers of Lateral Lymph Node Metastasis
-
Mijin Kim1
 , Chae Hwa Kwon2, Min Hee Jang1, Jeong Mi Kim1, Eun Heui Kim1, Yun Kyung Jeon1, Sang Soo Kim1, Kyung-Un Choi3, In Joo Kim1, Meeyoung Park4, Bo Hyun Kim1
, Chae Hwa Kwon2, Min Hee Jang1, Jeong Mi Kim1, Eun Heui Kim1, Yun Kyung Jeon1, Sang Soo Kim1, Kyung-Un Choi3, In Joo Kim1, Meeyoung Park4, Bo Hyun Kim1 -
Endocrinology and Metabolism 2021;36(5):1086-1094.
DOI: https://doi.org/10.3803/EnM.2021.1132
Published online: October 28, 2021
1Department of Internal Medicine, Biomedical Research Institute, Pusan National University Hospital, Busan, Korea
2Biomedical Research Institute, Pusan National University Hospital, Busan, Korea
3Department of Pathology, Biomedical Research Institute, Pusan National University Hospital, Busan, Korea
4Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, Korea
- Corresponding authors: Bo Hyun Kim. Department of Internal Medicine, Biomedical Research Institute, Pusan National University Hospital, 305 Gudeok-ro, Seo-gu, Busan 49241, Korea Tel: +82-51-240-7236, Fax: +82-51-254-3237, E-mail: pons71@pusan.ac.kr
- Meeyoung Park. Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, 20 Geumo-ro, Mulgeum-eup, Yangsan 50612, Korea Tel: +82-55-360-4735, Fax: +82-55-360-3869, E-mail: mpark.pnuh@gmail.com
Copyright © 2021 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Early identification of patients with high-risk papillary thyroid microcarcinoma (PTMC) that is likely to progress has become a critical challenge. We aimed to identify somatic mutations associated with lateral neck lymph node (LN) metastasis (N1b) in patients with PTMC.
-
Methods
- Whole-exome sequencing (WES) of 14 PTMCs with no LN metastasis (N0) and 13 N1b PTMCs was performed using primary tumors and matched normal thyroid tissues.
-
Results
- The mutational burden was comparable in N0 and N1b tumors, as the median number of mutations was 23 (range, 12 to 46) in N0 and 24 (range, 12 to 50) in N1b PTMC (P=0.918). The most frequent mutations were detected in PGS1, SLC4A8, DAAM2, and HELZ in N1b PTMCs alone, and the K158Q mutation in PGS1 (four patients, Fisher’s exact test P=0.041) was significantly enriched in N1b PTMCs. Based on pathway analysis, somatic mutations belonging to the receptor tyrosine kinase-RAS and NOTCH pathways were most frequently affected in N1b PTMCs. We identified four mutations that are predicted to be pathogenic in four genes based on Clinvar and Combined Annotation-Dependent Depletion score: BRAF, USH2A, CFTR, and PHIP. A missense mutation in CFTR and a nonsense mutation in PHIP were detected in N1b PTMCs only, although in one case each. BRAF mutation was detected in both N0 and N1b PTMCs.
-
Conclusion
- This first comprehensive WES analysis of the mutational landscape of N0 and N1b PTMCs identified pathogenic genes that affect biological functions associated with the aggressive phenotype of PTMC.
- The incidence of thyroid cancer has increased worldwide, and most of these newly diagnosed cancers are papillary thyroid microcarcinomas (PTMCs), defined as tumors of size ≤1 cm [1]. Although most PTMCs exhibit an indolent clinical course, some cases show an aggressive phenotype with poor prognosis [2]. It is not clear which PTMCs require more aggressive treatments and more careful surveillance. Therefore, at the time of diagnosis, the identification of patients with high-risk PTMC that is likely to progress has become one of the most important challenges in the management of PTMC. To enable early assessment of patients with high-risk PTMC, it is crucial to discover the pathobiological mechanisms that govern the initiation of PTMC metastasis and tumor growth.
- Lateral neck lymph node (LN) metastasis (N1b) is a representative risk factor for disease progression, aggressive tumor behavior, high disease recurrence, and mortality [3-5]. Although it is known that LN metastasis can be caused by unique molecular alterations, studies on the mutation profiles of PTMC with N1b are lacking [6,7]. A recent study reported differential RNA expression of 43 genes between PTMCs with N1b and PTMCs without nodal disease (N0) by using a custom panel of 248 RNAs [6]. In that study, the mutations found in the telomerase reverse transcriptase (TERT) promoter and in TP53 were exclusive to N1b cases [6]. Another study performed targeted nextgeneration sequencing (NGS) using a panel of 382 genes in PTMCs with metastatic LNs, demonstrating that the mutational status of the primary tumors did not significantly differ from that of the corresponding metastatic LN [7]. Both studies performed targeted sequencing analysis using customized NGS panels, and the biological role of the identified genomic alterations is still under examination.
- Whole-exome sequencing (WES) is an effective approach to identify cancer mutations in coding regions of the genome, allowing for the identification of potential causative alterations in as-yet unknown genes [8]. WES can provide more comprehensive data than can be achieved through targeted sequencing panels [9,10]. To date, WES has allowed the identification of genomic mutation profiles in several cancer types [11-13]. However, to the best of our knowledge, no studies have employed WES to compare low-risk (N0) and high-risk (N1b) PTMCs.
- Thus, we hypothesized that WES analysis of the primary tumors could identify novel key genes associated with the aggressive phenotype that is observed in a subset of PTMCs. We performed WES on normal and tumor DNA pairs in 14 N0 and 13 N1b patients to compare the mutational profiles in N0 and N1b PTMCs and discover pathogenic genes potentially contributing to the aggressive tumor phenotype.
INTRODUCTION
- Patients and sample collection
- This study included patients with PTMC (14 N0 and 13 N1b) who underwent initial thyroid surgery at the Pusan National University Hospital, Busan, Korea. The biospecimens and data used for this study were provided by the Biobank of the Pusan National University Hospital, a member of the Korea Biobank Network. Tumor and the matched normal fresh frozen tissues were collected through several steps by pathologists. In detail, after gross examination, non-necrotic portions were excised from the resected tumor specimens by pathologists and were immersed in liquid nitrogen immediately. Matched normal thyroid tissue was obtained 2 cm away from the tumor. Sample adequacy for tumor content was assessed at the time of procurement and again on processed blocks by an experienced pathologist (K.U.C.). All tumor samples used in this study passed the purity assessment, and the average tumor cell content was 65%. Written informed consent was obtained from all participants prior to the survey, and the protocol of this study was approved by the Institutional Review Board of the Pusan National University Hospital, Busan, Korea (No. H-1804-021-065).
- DNA preparation and sequencing
- DNA was extracted from fresh frozen tissue samples using the QuickGene DNA tissue kit S with a QG-Mini80 instrument (Kurabo, Osaka, Japan). SureSelect sequencing libraries were prepared according to the manufacturer’s instructions (Agilent SureSelectXT Human All Exon V5, Agilent, Santa Clara, CA, USA) by using the Bravo automated liquid handler. Sequencing was performed using an Illumina Novaseq 6000 system following the provided protocols for 2×100 sequencing at DNA Link Inc. (Seoul, Korea).
- Variant calling and data processing
- Quality assessment for the generated WES was performed using FastQC version 0.11.5 [14]. Burrows-Wheeler Aligner version 0.7.12 [15] was used for mapping and alignment based on Genome Reference Consortium Human Build 37 (GRCh37). Next, GATK version 3.5 was used for realignment and recalibration [16]. Variant calling for single nucleotide variants and indels was performed using MuTect version 1.1.7 [17] and Strelka version 1.0.14 [18]. Input sequence data from matched pairs for tumor and normal DNA were analyzed using MuTect/Strelka and, after removal of low-quality reads, the presence of variants in addition to expected random sequencing errors was determined [17,19]. Statistical analysis was performed to identify sites that may have a high probability for the occurrence of somatic mutations by calculating a log-odds score with high confidence. The score was compared to a cutoff determined using the log ratio of prior probabilities of the events under consideration. To reduce the risk of identification of false positives and remove artifacts, six different filters were used to screen candidate variant sites: (1) proximal gap; (2) poor mapping; (3) triallelic sites; (4) strand bias; (5) clustered positions; and (6) presence in the controls [17]. Somatic mutations were identified by comparing the sequencing data for matched tumor and normal samples, and we filtered germline mutations using the single nucleotide polymorphism (SNP) database build 134 and 1,000 Genome data. To reduce the occurrence of false positives, we filtered variants showing <5% variant allele frequency and with total coverage under 15×. Furthermore, missense SNVs and loss of function variants were used for subsequent analyses. The identified variants were annotated by Oncotator v1.8.0 [20] for functional information. Information on variant pathogenicity was annotated using ClinVar and Combined Annotation-Dependent Depletion (CADD) [21]. Variants with CADD scores over 30, which are predicted to be the 0.1% most deleterious substitutions possible in the human genome [21], were defined as pathogenic mutations.
- Statistical analysis
- Statistical analyses were performed by using the R program version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria). Continuous variables are presented as medians with interquartile range (IQR) and were analyzed by Student’s t test. Categorical variables are presented as frequencies and expressed as percentages, and they were analyzed by chi-square test. The MafCompare function in the Maftools package was used to determine associations between differentially mutated genes and lateral neck disease, wherein the mutation load for each gene was compared by Fisher’s exact test [22]. To compare the somatic mutation profiles of papillary thyroid carcinoma (PTC) using The Cancer Genome Atlas (TCGA), we obtained WES data including information on primary tumor sizes and LN metastasis from 291 patients [23]. P<0.05 was considered statistically significant.
METHODS
- Baseline characteristics
- The baseline characteristics of the 14 N0 and 13 N1b PTMCs are listed in Table 1. All patients had classical PTC. The median age of the patients at the time of initial thyroidectomy was 50 years (IQR, 46 to 61), and 23 (85%) patients were female. The median primary tumor size was 0.9 cm (IQR, 0.7 to 1.0), with no significant differences between the N0 and the N1b group (P=0.871). Microscopic and gross extra-thyroidal extension (ETE) were identified in 16 (59%) and two (7%) patients, respectively. Six (22%) patients had multifocal PTC, and lymphovascular invasion was present in two (7%) patients.
- All patients underwent prophylactic central compartment node dissection, whereas therapeutic central and lateral neck dissection was performed in patients with clinically apparent lateral cervical LN metastasis. The median number of removed LNs was 4 (IQR, 2 to 8) in N0 and 35 (IQR, 32 to 50) in N1b PTMCs. Among the removed LNs, the median number of metastatic LNs was 7 (IQR, 3 to 15), with a maximum size of 12 mm (IQR, 7 to 12). Among patients with N1b, extranodal extension of metastatic foci from the metastatic LNs was detected in five cases.
- Whole-exome sequencing and identified somatic mutations
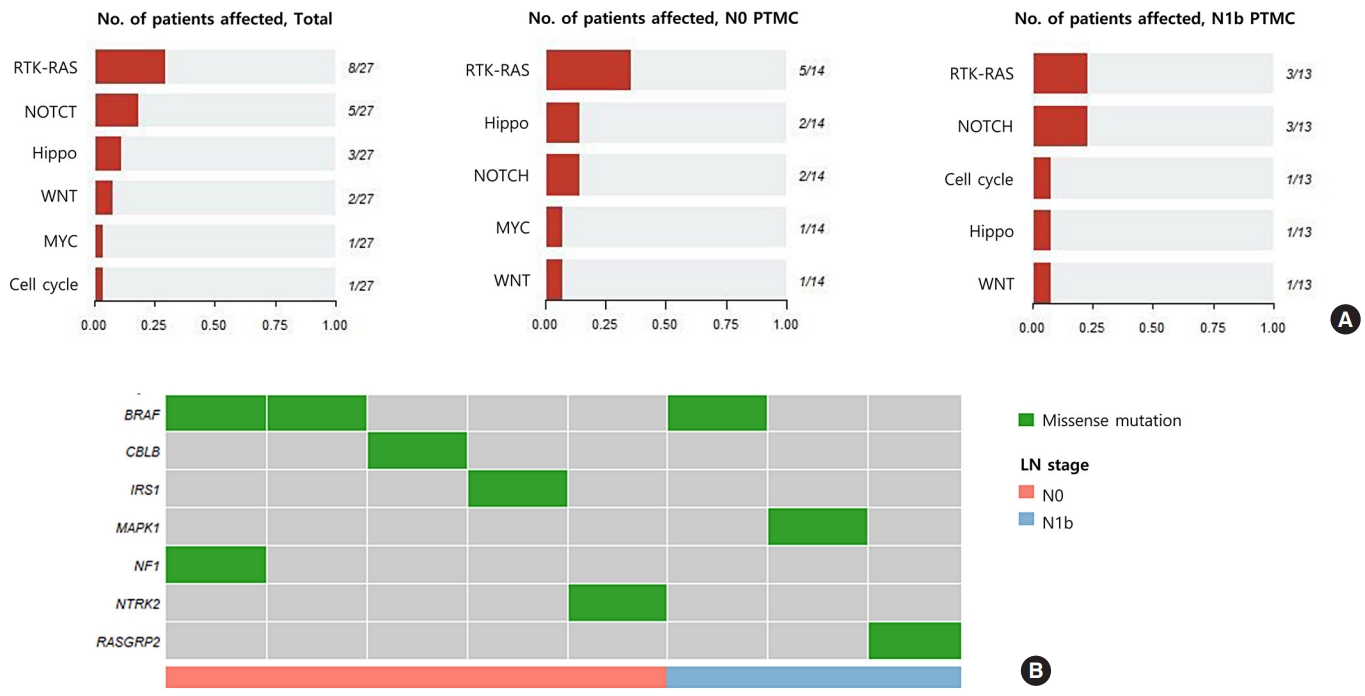
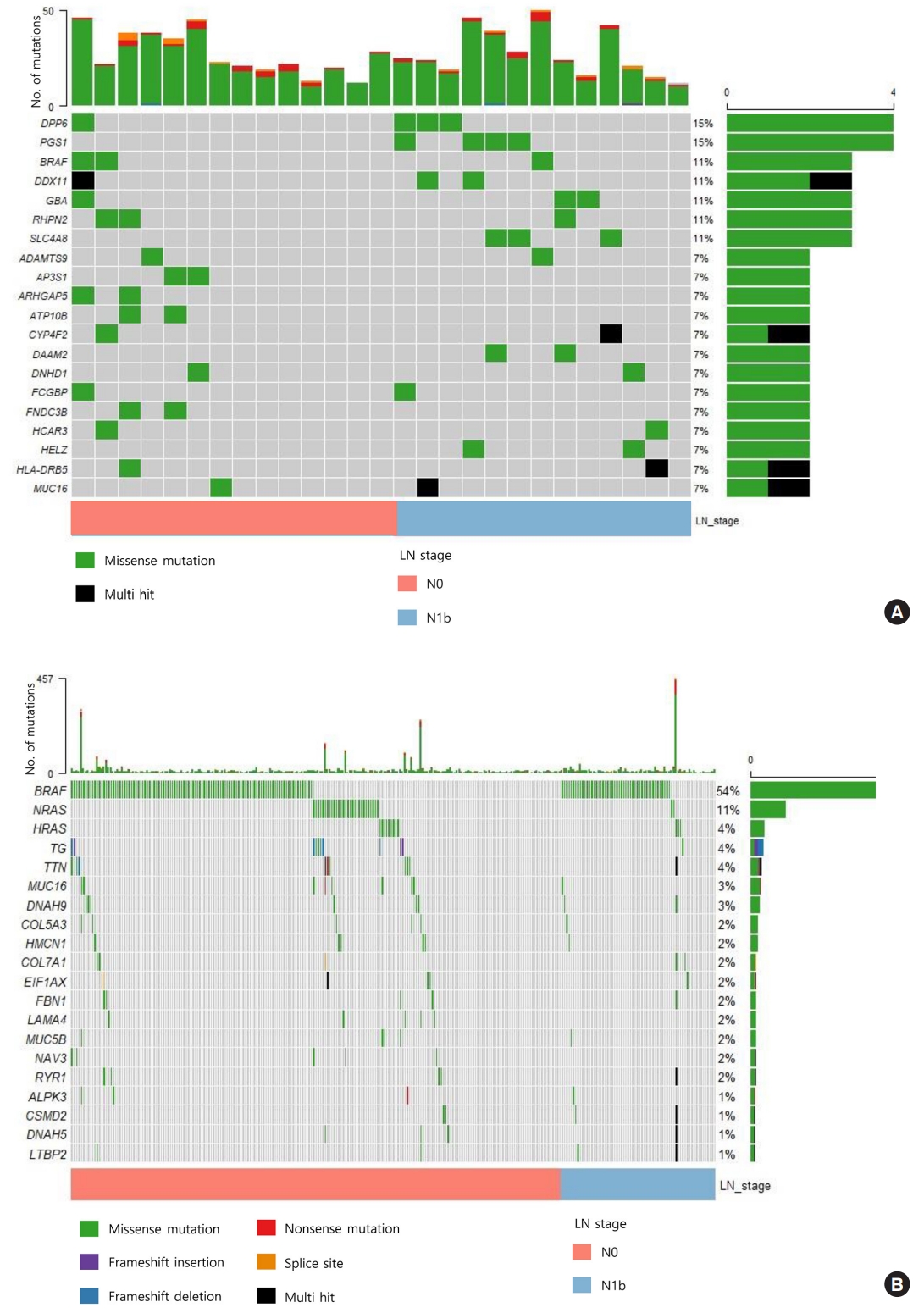
- We sequenced 27 matched DNA sample pairs from primary tumors and normal thyroid tissues. WES was performed to 100× coverage at the raw data level, and a median target coverage depth of 67× (range, 53× to 91×) in primary tumors and 70× (range, 50× to 99×) in matched normal thyroid tissues was followed (Supplemental Table S1). After calling variants with MuTect and Strelka, with highly sensitive algorithms and specific cutting-edge mutation calling methods, we identified 313 and 281 significant somatic variants in N0 and N1b tumors, respectively. The mutation burden was not significantly different between N0 and N1b tumors, with a median of 23 (range, 12 to 46) and 24 mutations (range, 12 to 50) in N0 and N1b PTMCs, respectively (P=0.918) (Supplemental Fig. S1). The top 20 genomic alterations across cases are shown in Fig. 1A, Supplemental Table S2. The most frequently mutated genes were DPP6, and PGS1, each occurring in four cases. Mutations in PGS1 SLC4A8, DAAM2 and HELZ were detected only in N1b disease, and the K158Q mutation in PGS1 was significantly enriched in N1b PTMCs (four cases; Fisher’s exact test P=0.041). BRAF mutations, all represented by the V600E mutation, were detected in three PTMCs (11%), i.e., two N0 (14%) tumors and one N1b (8%) tumor. Somatic alterations in genes included those that are a part of the receptor tyrosine kinase (RTK)-RAS signaling pathway; mutations in BRAF, CBLC, IRS1, NF1, and NTRK2 in N0 and BRAF, MAPK1, and RASGRP2 in N1b were most frequently detected in patients with PTMC (Fig. 2). The RTK-RAS and NOTCH pathways were impacted in three N1b patients. In the TCGA dataset, the BRAF V600E mutation was the most frequent alteration (54%), and was detected more frequently in N1b (66%) than N0 PTCs (49%, P=0.002) (Fig. 1B). The NRAS mutation was identified in 11% of patients with PTC, and the frequency of detection was higher in N0 (13%) than N1b tumors (3%, P=0.013). The BRAF V600E mutation was confirmed via polymerase chain reaction (PCR)-based Sanger sequencing (Supplemental Fig. S2).
- Pathogenic mutations in N0 and N1b PTMCs
- To identify mutations implicated in the aggressive PTMC phenotype, we evaluated pathogenic mutations using ClinVar and the CADD scores (>30). We identified a median of 3 (range, 1 to 11) pathogenic mutations per case among N0 PTMCs and 4 (range, 2 to 7) among N1b PTMCs (Supplemental Tables S3, S4). Among them, BRAF, USH2A, CFTR, and PHIP mutations were classified as pathogenic based on both ClinVar and CADD scores (Table 2). In N0 PTMC, the BRAF V600E mutation (two cases) and a nonsense mutation in USH2A (one case) were detected. In N1b PTMC, the V600E mutation in BRAF (one case), a missense mutation in CFTR (one case), and nonsense mutations in USH2A (one case) and PHIP (one case) were detected. The CFTR V561E mutated N1b PTMC showed microscopic ETE with histological features of Hashimoto’s thyroiditis. Another single N1b PTMC harbored a nonsense mutation in PHIP (p.Y235*) and showed microscopic ETE and lymphovascular invasion. Both tumors were multifocal PTMCs. In the TCGA dataset, one N0 PTC harbored a missense mutation in CFTR (p.L702I) and there were no tumors that showed mutations in PHIP.
RESULTS
- In this study, we used WES to identify important genomic alterations that might impact the aggressiveness of PTMC. Although the mutational burden did not vary between N0 and N1b PTMCs, the mutation profiles were distinct in the two groups of tumors. The most frequent mutations were detected in PGS1, SLC4A8, DAAM2, and HELZ in N1b PTMCs alone, and the K158Q mutation in PGS1 was significantly enriched in N1b PTMCs. Pathway analysis showed that somatic mutations in the RTK-RAS pathway were most frequent, and these results supported those from studies using TCGA data which identified a on in PGS1 was signcritical role for the MAPK pathway alterations in PTC [23]. In N1b PTMCs, the RTK-RAS and NOTCH pathways were affected in 19% of patients. We identified four mutations that are predicted to be pathogenic in four genes: BRAF, USH2A, CFTR, and PHIP. Mutations in BRAF (two in N0 and one in N1b) and USH2A (one each in N0 and N1b) were detected in both N0 and N1b PTMCs. However, CFTR and PHIP mutations were differentially present in N1b, although only in one case. These results suggested that mutation may be heterogeneously distributed in PTMCs. Thus, further research is needed on whether mutations in BRAF and USH2A are acquired at the early stage during tumor progression and whether mutations of CFTR and PHIP are specific to LN metastasis.
- The presence of the BRAF V600E mutation was not predictive of clinically significant lateral neck LN metastasis. This mutation plays an important oncogenic role in thyroid tumorigenesis through the activation of the MAPK pathway [24]. Notably, most studies have shown that aggressive behavior is observable in a subset of PTMCs harboring the BRAF V600E mutation [25,26]. However, in accordance with our study, a recent NGS-based genomic analysis demonstrated that BRAF V600E alterations are present at approximately equal frequencies in N0 and N1b PTMCs [6]. The results from our study and the previous study [6], which were specifically designed to evaluate differences in genomic alterations between N0 and N1b PTMCs, indicate that testing for BRAF V600E mutation alone has limited value in predicting the development of LN metastasis in patients with PTMC.
- Alterations in the CFTR gene can cause cystic fibrosis, which affects the epithelial tissues of various organs, including the respiratory, gastrointestinal, and urogenital system [27,28]. Subclinical hypothyroidism associated with cystic fibrosis caused by iodine fluxes is known to be mediated by CFTR in the thyroid [27]. Cystic fibrosis is also correlated with an increased risk of thyroid cancer [29]. Oh et al. [30] reported that in PTC, two SNPs (rs4148682, promoter; rs213950, p.V470M) in CFTR have prognostic significance and may be associated with clinical features such as multifocality, cancer location, and cervical LN metastasis. In the TCGA database, the missense L702I mutation in CFTR was detected in a multifocal PTC (3.2 cm in the great dimension) along with N0 tumors. In this study, the missense SNP (rs121909047, p.A561E) in CFTR was detected in a PTMC with N1b tumors. Additional analysis is required to verify the association between the A561E mutation in CFTR, aggressive phenotypes, and prognosis of PTMC.
- A nonsense mutation in PHIP (p.Y235*) was detected in one N1b PTMC with an aggressive phenotype. PHIP has a WD40 repeat and two bromodomains and is expressed in a wide range of tissues including the thyroid gland. A recent in vivo study identified a role for PHIP as a potent tumor suppressor in B-cell lymphomagenesis [31]. The authors found that mice transduced with a PHIP sgRNA showed significantly accelerated B-cell lymphoma development. In addition, B-cell lymphomas harvested from these animals showed significantly reduced PHIP expression. To our knowledge, no studies have investigated the molecular function or clinical impact of PHIP mutations on thyroid cancer. Additional studies are required to characterize the role of PHIP mutations as tumor suppressors in thyroid cancer in detail.
- A nonsense mutation in USH2A (c8271T>G, p.Y2757*) was detected in both N0 and N1b PTMCs. It is different from the missense mutations (I2189V, D798V, E4571K, and L1727F) in USH2A that were reported in anaplastic thyroid cancer [32]. This gene encodes a protein called usherin, an important component of basement membranes [33], and is one of the top 10 mutated genes across various human tumor types [34]. Alterations in the extracellular matrix or cellular adhesion functions can lead to progression to highly invasive and metastatic tumors.
- The most significant difference relative to the TCGA data was a relatively low prevalence of the BRAF V600E mutation. The frequency of BRAF mutated tumor in our study is also lower than that of previously reported (29% to 83%) [24]. There are a few possible reasons for this discrepancy, including insufficient sequencing depth. Regarding the coverage depth of WES, PCR-based Sanger sequencing was performed on all tumor samples to overcome the insufficient sequencing depth. We only detected the BRAF V600E mutation in three cases, Case 2, Case 14, and Case 20 (Supplemental Fig. S2), as in the WES results. These results suggest the possibility of identifying driver mutations by WES analysis despite low coverage depth. Therefore, regarding the discrepancy in the frequency of BRAF mutation in thyroid cancer, we need to consider a variety of issues, including patient clinical characteristics, histological type of samples, sample number, and detection methods, as compared with other studies. The limited number of samples and a high proportion of indolent tumors in our study cohort may cause this discrepancy.
- Our study has several limitations. First, as the metastatic LN tissue samples were difficult to obtain from the enrolled patients, we evaluated the mutational profiles of primary tumors only. The use of matched normal thyroid tissue as a normal DNA control, rather than matched leukocyte DNA or other matched normal DNA, also likely affected the mutation profiles. Second, exome sequencing is limited in detecting copy number variations and structural rearrangement. Further, the Agilent SureSelect V5 capture does not capture the TERT promoter region. Nonetheless, it should be taken into account that some of the identified factors may be potential early predictors of PTMC progression.
- This first comprehensive WES analysis of the mutational landscape of N0 and N1b PTMCs identified pathogenic genes potentially associated with the aggressive PTMC phenotype. These results may be clinically important in identifying patients who may benefit from radioactive iodine treatment or completion of thyroidectomy if not performed at the time of diagnosis. Future studies are needed to clarify the molecular role of these alterations in the development of lateral neck LN metastasis in PTMC.
DISCUSSION
Supplementary Information
Supplemental Table S3.
Supplemental Table S4.
Supplemental Fig. S1.
Supplemental Fig. S2.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: M.K., C.H.K., M.P., B.H.K. Acquisition, analysis, or interpretation of data: M.K., C.H.K., M.H.J., J. M.K., E.H.K., Y.K.J., S.S.K., K.U.C., I.J.K., M.P., B.H.K. Drafting the work or revision: M.K., C.H.K, M.P., B.H.K. Final approval of the manuscript: M.K., C.H.K., M.H.J., J.M.K., E.H.K., Y.K.J., S.S.K., K.U.C., I.J.K., M.P., B.H.K.
Article information
-
Acknowledgements
- This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF-2018M3A9E8066253). This study was supported by Biomedical Research Institute Grant (20210019), Pusan National University Hospital. The biospecimens and data used for this study were provided by the Biobank of Pusan National University Hospital, a member of the Korea Biobank Network.

- 1. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016;26:1–133.ArticlePubMedPMC
- 2. Li D, Gao M, Li X, Xing M. Molecular aberrance in papillary thyroid microcarcinoma bearing high aggressiveness: identifying a “Tibetan Mastiff dog” from puppies. J Cell Biochem 2016;117:1491–6.ArticlePubMed
- 3. Nixon IJ, Wang LY, Palmer FL, Tuttle RM, Shaha AR, Shah JP, et al. The impact of nodal status on outcome in older patients with papillary thyroid cancer. Surgery 2014;156:137–46.ArticlePubMed
- 4. Mercante G, Frasoldati A, Pedroni C, Formisano D, Renna L, Piana S, et al. Prognostic factors affecting neck lymph node recurrence and distant metastasis in papillary microcarcinoma of the thyroid: results of a study in 445 patients. Thyroid 2009;19:707–16.ArticlePubMed
- 5. Pisanu A, Saba A, Podda M, Reccia I, Uccheddu A. Nodal metastasis and recurrence in papillary thyroid microcarcinoma. Endocrine 2015;48:575–81.ArticlePubMed
- 6. Perera D, Ghossein R, Camacho N, Senbabaoglu Y, Seshan V, Li J, et al. Genomic and transcriptomic characterization of papillary microcarcinomas with lateral neck lymph node metastases. J Clin Endocrinol Metab 2019;104:4889–99.ArticlePubMedPMC
- 7. Jeon MJ, Chun SM, Lee JY, Choi KW, Kim D, Kim TY, et al. Mutational profile of papillary thyroid microcarcinoma with extensive lymph node metastasis. Endocrine 2019;64:130–8.ArticlePubMed
- 8. Lee WK, Lee SG, Yim SH, Kim D, Kim H, Jeong S, et al. Whole exome sequencing identifies a novel hedgehog-interacting protein G516R mutation in locally advanced papillary thyroid cancer. Int J Mol Sci 2018;19:2867.ArticlePubMedPMC
- 9. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–55.ArticlePubMed
- 10. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014;370:2418–25.ArticlePubMed
- 11. Masoodi T, Siraj AK, Siraj S, Azam S, Qadri Z, Albalawy WN, et al. Whole-exome sequencing of matched primary and metastatic papillary thyroid cancer. Thyroid 2020;30:42–56.ArticlePubMedPMC
- 12. Miller AL, Garcia PL, Pressey JG, Beierle EA, Kelly DR, Crossman DK, et al. Whole exome sequencing identified sixty-five coding mutations in four neuroblastoma tumors. Sci Rep 2017;7:17787.ArticlePubMedPMC
- 13. Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer 2016;55:251–67.ArticlePubMed
- 14. Andrews S. FastQC: A quality control tool for high throughput sequence data [Internet]. Cambridge: Babraham Bioinformatics; 2010 [cited 2021 Sep 23]. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 15. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95.ArticlePubMedPMC
- 16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303.ArticlePubMedPMC
- 17. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9.ArticlePubMedPMC
- 18. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018;15:591–4.ArticlePubMed
- 19. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7.ArticlePubMed
- 20. Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, et al. Oncotator: cancer variant annotation tool. Hum Mutat 2015;36:E2423–9.ArticlePubMedPMC
- 21. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5.ArticlePubMedPMC
- 22. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 2018;28:1747–56.ArticlePubMedPMC
- 23. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159:676–90.PubMedPMC
- 24. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer 2005;12:245–62.ArticlePubMed
- 25. Nucera C, Pontecorvi A. Clinical outcome, role of BRAF (V600E), and molecular pathways in papillary thyroid microcarcinoma: is it an indolent cancer or an early stage of papillary thyroid cancer? Front Endocrinol (Lausanne) 2012;3:33.ArticlePubMedPMC
- 26. Choi SY, Park H, Kang MK, Lee DK, Lee KD, Lee HS, et al. The relationship between the BRAF(V600E) mutation in papillary thyroid microcarcinoma and clinicopathologic factors. World J Surg Oncol 2013;11:291.ArticlePubMedPMC
- 27. Li H, Ganta S, Fong P. Altered ion transport by thyroid epithelia from CFTR(-/-) pigs suggests mechanisms for hypothyroidism in cystic fibrosis. Exp Physiol 2010;95:1132–44.ArticlePubMedPMCPDF
- 28. Wilschanski M, Durie PR. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut 2007;56:1153–63.ArticlePubMedPMC
- 29. Johannesson M, Askling J, Montgomery SM, Ekbom A, Bahmanyar S. Cancer risk among patients with cystic fibrosis and their first-degree relatives. Int J Cancer 2009;125:2953–6.ArticlePubMed
- 30. Oh IH, Oh C, Yoon TY, Choi JM, Kim SK, Park HJ, et al. Association of CFTR gene polymorphisms with papillary thyroid cancer. Oncol Lett 2012;3:455–61.ArticlePubMed
- 31. Weber J, de la Rosa J, Grove CS, Schick M, Rad L, Baranov O, et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat Commun 2019;10:1415.ArticlePubMedPMC
- 32. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet 2015;24:2318–29.ArticlePubMedPMC
- 33. Bhattacharya G, Kalluri R, Orten DJ, Kimberling WJ, Cosgrove D. A domain-specific usherin/collagen IV interaction may be required for stable integration into the basement membrane superstructure. J Cell Sci 2004;117(Pt 2):233–42.Article
- 34. Kim N, Hong Y, Kwon D, Yoon S. Somatic mutaome profile in human cancer tissues. Genomics Inform 2013;11:239–44.ArticlePubMedPMC
References
Figure & Data
References
Citations
- What can we learn about acid-base transporters in cancer from studying somatic mutations in their genes?
Bobby White, Pawel Swietach
Pflügers Archiv - European Journal of Physiology.2024; 476(4): 673. CrossRef - Comprehensive Long-Read Sequencing Analysis Discloses the Transcriptome Features of Papillary Thyroid Microcarcinoma
Yanqiang Wang, Binbin Zou, Yanyan Zhang, Jin Zhang, Shujing Li, Bo Yu, Zhekun An, Lei Li, Siqian Cui, Yutong Zhang, Jiali Yao, Xiuzhi Shi, Jing Liu
The Journal of Clinical Endocrinology & Metabolism.2024; 109(5): 1263. CrossRef - Feasibility of whole‐exome sequencing in fine‐needle aspiration specimens of papillary thyroid microcarcinoma for the identification of novel gene mutations
Liyuan Ma, Luying Gao, Ya Hu, Xiaoyi Li, Chunhao Liu, Jiang Ji, Xinlong Shi, Aonan Pan, Yuang An, Nengwen Luo, Yu Xia, Yuxin Jiang
Clinical Genetics.2024; 105(5): 567. CrossRef - Multi-omics analysis reveals a molecular landscape of the early recurrence and early metastasis in pan-cancer
Dan-ni He, Na Wang, Xiao-Ling Wen, Xu-Hua Li, Yu Guo, Shu-heng Fu, Fei-fan Xiong, Zhe-yu Wu, Xu Zhu, Xiao-ling Gao, Zhen-zhen Wang, Hong-jiu Wang
Frontiers in Genetics.2023;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite