Docosahexanoic Acid Attenuates Palmitate-Induced Apoptosis by Autophagy Upregulation via GPR120/mTOR Axis in Insulin-Secreting Cells
Article information

Abstract
Background
Polyunsaturated fatty acids (PUFAs) reportedly have protective effects on pancreatic β-cells; however, the underlying mechanisms are unknown.
Methods
To investigate the cellular mechanism of PUFA-induced cell protection, mouse insulinoma 6 (MIN6) cells were cultured with palmitic acid (PA) and/or docosahexaenoic acid (DHA), and alterations in cellular signaling and apoptosis were examined.
Results
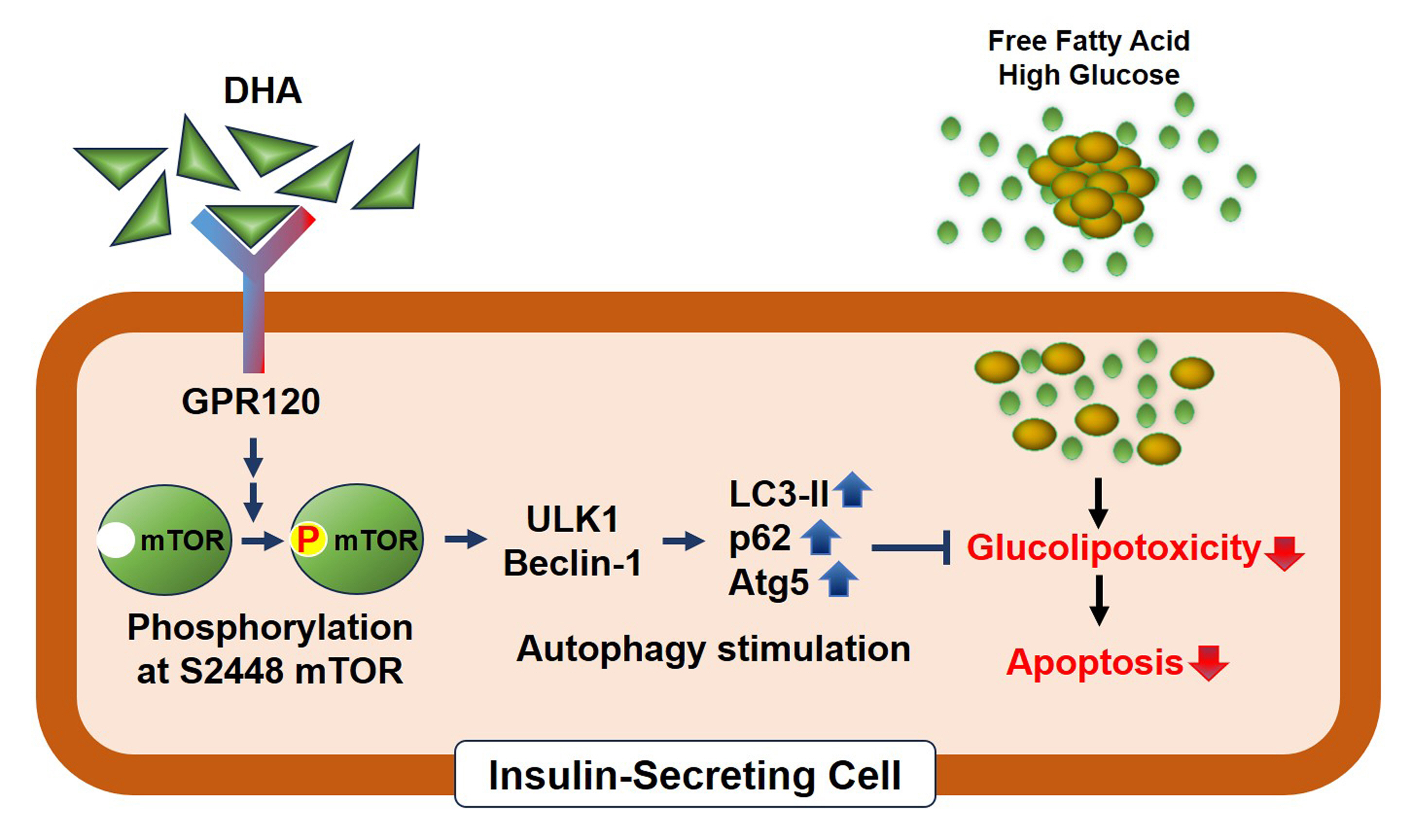
DHA treatment remarkably repressed caspase-3 cleavage and terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL)-positive red dot signals in PA-treated MIN6 cells, with upregulation of autophagy, an increase in microtubule-associated protein 1-light chain 3 (LC3)-II, autophagy-related 5 (Atg5), and decreased p62. Upstream factors involved in autophagy regulation (Beclin-1, unc51 like autophagy activating kinase 1 [ULK1], phosphorylated mammalian target of rapamycin [mTOR], and protein kinase B) were also altered by DHA treatment. DHA specifically induced phosphorylation on S2448 in mTOR; however, phosphorylation on S2481 decreased. The role of G protein-coupled receptor 120 (GPR120) in the effect of DHA was demonstrated using a GPR120 agonist and antagonist. Additional treatment with AH7614, a GPR120 antagonist, significantly attenuated DHA-induced autophagy and protection. Taken together, DHA-induced autophagy activation with protection against PA-induced apoptosis mediated by the GPR120/mTOR axis.
Conclusion
These findings indicate that DHA has therapeutic effects on PA-induced pancreatic β-cells, and that the cellular mechanism of β-cell protection by DHA may be a new research target with potential pharmacotherapeutic implications in β-cell protection.
INTRODUCTION
Polyunsaturated fatty acids (PUFAs), including ω-3 and ω-6, are pivotal components in the regulation of cellular lipid metabolism. Constant supplementation with ω-3, such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), is known to be beneficial for maintaining health. Indeed, reduced ω-3 levels are related to metabolic disorders [1,2], and individuals with higher levels of ω-3 PUFAs have a lower risk of type 2 diabetes [3].
Dysfunctional pancreatic β-cells and abdominal obesity are the major causes of type 2 diabetes. They can also be a crucial treatment target [4,5]. Saturated non-esterified fatty acids (NEFAs) are harmful to pancreatic β-cells. Chronic exposure to elevated NEFA and glucose levels strongly induces the dysfunction and apoptosis of pancreatic β-cells [6-8]. However, several studies have suggested that unsaturated fatty acids exert beneficial effects on pancreatic β-cells, despite the toxicity of NEFAs. A previous study has reported that treatment with ω-3 protects pancreatic islets against NEFA-induced cell disorders [9]. Moreover, Bellini et al. [10] showed that de novo synthesis of DHA, an ω-3 PUFA, is essential for maintaining cell survival and inhibiting glucolipotoxicity-induced cell death in pancreatic β-cells. These studies demonstrated the protective role of DHA from dietary supplements and endogenous synthesis; however, the underlying mechanisms and related cellular pathways are unknown.
In recent studies, a family of G protein-coupled receptors (GPRs), which are activated by unsaturated fatty acids, was identified and characterized as a target to cure diabetes [11]. Among GPRs, GPR40 and 120 are regulated by long-chain fatty acids, especially DHA and EPA [12,13], and GPR120 has been indicated to have a cell-protective role via its anti-inflammatory effect in various cells, including hepatocytes [14], neurons [15], and macrophages [16]. In particular, GPR120 has been proposed to improve β-cell function and insulin resistance. A previous study showed that genetic deletion of GPR120 resulted in impaired glucose tolerance, with reduced insulin content in the islets compared to those of wild-type littermates [17], indicating that GPR120 is crucial for maintaining β-cell homeostasis.
Autophagy is an intracellular self-recycling process that involves the degradation of unnecessary or damaged organelles [18]. Autophagy is essential for maintaining cellular homeostasis and cell survival under stressful conditions such as nutritional imbalance (e.g., starvation or glucolipotoxicity) [19]. Autophagy is particularly critical in pancreatic β-cells because of their ability to synthesize insulin. Indeed, remarkable upregulation of autophagic flux was observed in β-cells from mice fed a high-fat diet and in mouse insulinoma 6 (MIN6) cells treated with NEFAs [20,21]. In addition, β-cell-specific ablation of autophagy-related 7 (Atg7), an essential autophagy gene, induces progressive β-cell loss and dysfunction leading to hyperglycemia, and pharmacological facilitation of autophagy improves β-cell survival and function [22], suggesting that autophagy plays a pivotal role in maintaining β-cell homeostasis.
Although the beneficial function of PUFAs has been identified in many organs and cells, the underlying mechanism of action in insulin-secreting cells has not been investigated. In the present study, we aimed to estimate the molecular mechanism of the protective effect induced by DHA by targeting autophagy and the related cellular signaling pathways in insulin-secreting cells.
METHODS
Cell culture and treatment
MIN6 cell lines (passages 16–22) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) containing high glucose (4.5 g/L) supplemented with heat-inactivated 10% fetal bovine serum, 100 IU/mL penicillin, 50 μg/mL streptomycin, and 50 μM 2-mercaptoethanol (Life Technologies Corporation, Paisley, UK). Cells were preincubated with serum-starved medium for 2 hours, followed by incubation in medium containing DHA (40 μM; Merck, Darmstadt, Germany), palmitic acid (PA, 500 μM; R&D Systems Inc., Minneapolis, MN, USA), or a combination of both for 24 hours. Cells were preincubated with AH7614 (20 μM; Tocris Bioscience, Bristol, UK) or GSK137647 (50 μM; Tocris Bioscience) before treatment with DHA and/or PA.
In situ detection of fragmented DNA (TUNEL) assay
DNA fragmentation was examined using a commercial kit to detect apoptosis (ApopTag Red; Merck, Temecula, CA, USA). After fixation and permeabilization, cells were incubated with 75 μL equilibration buffer for 3 minutes and then exposed to 55 μL working strength terminal transferase enzyme solution in a humidified chamber at 37°C for 1 hour. After washing with stop solution and washing buffer, the cells were incubated with warmed anti-digoxigenin conjugate solution at 37°C in a dark chamber. The cells were then counterstained with 4’,6-diamidino-2-phenylindole (DAPI, Invitrogen, Carlsbad, CA, USA) for 5 minutes and mounted on slides after washing with phosphatebuffered saline (Gibco, Bleiswjik, Netherlands).
Immunoblotting analysis
Protein expression levels were evaluated by immunoblotting analysis as previously described [23]. Briefly, whole cell lysates (WCLs) were extracted using radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and protease/phosphatase inhibitor (Thermo Fisher Scientific, Waltham, MA, USA). The prepared WCLs were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The following primary antibodies were used: anti-cleaved caspase-3, anti-total caspase-3, anti-β-actin, anti-light chain 3 (LC3)-I/II, anti-p62, anti-Atg5, anti-Unc51 like autophagy activating kinase 1 (ULK1), anti-Beclin-1 (Abcam, Cambridge, MA, USA), anti-total mammalian target of rapamycin (mTOR), anti-phosphor-mTOR1 (p-2448S), anti-phosphor-mTOR2 (p2481S), antitotal-protein kinase B (Akt), anti-phosphor-Akt, anti-raptor, and anti-rictor (Cell Signaling Technology, Danvers, MA, USA).
Immunofluorescence analysis
We used a glass bottom dish (SPL Life Science, Pocheon, Korea) for the immunofluorescence assay, at 40% to 50% confluency; cells were incubated in completed medium in the presence and absence of DHA and/or PA as previously described. After removing the medium, the cells were washed and fixed in ice-cold 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) for 10 minutes. Then, cells were permeabilized in 0.01 M phosphate-buffered saline containing 0.5% Triton X-100 (Sigma-Aldrich) for 10 minutes. To reduce background, cells were incubated with 30% normal goat serum (Sigma-Aldrich) for 30 minutes then exposed to primary monoclonal antibody for LC3 (1:200; Abcam) overnight at 4°C. Fluorescein isothiocyanate (FITC) Alexa fluor anti-rabbit antibody (1:400; Zymed, Carlsbad, CA, USA) was used as a secondary antibody for 1 hour at 25°C. To estimate the co-localization of LC3 and Atg5, a double immunofluorescence assay was performed. Primary antibodies raised from different species were used to prevent non-specific binding, and target proteins were detected using the corresponding secondary antibodies conjugated with FITC or tetramethylrhodamine. To detect the nuclei, the cells were exposed to DAPI; the detected proteins were observed and analyzed using an Olympus BX51 microscope (Olympus Optical Co., Tokyo, Japan).
Statistical analysis
Statistical data are expressed as the mean±standard error of the mean. All calculations were performed using SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). For evaluation of statistical differences, Student’s t-test or one-way analysis of variance was performed, and multiple comparisons among the experimental groups were performed with the Bonferroni correction test. Differences with a P value <0.05 were considered significant.
RESULTS
DHA protects insulin-secreting cells against apoptosis by PA
Detrimental effects of NEFAs on pancreatic β-cells have also been reported. Chronic exposure to PA, a saturated NEFA, induces intracellular disorders, including endoplasmic reticulum stress [24], reactive oxygen species generation [23], ceramide accumulation, and impaired mitochondrial function [25], ultimately resulting in cell death. To investigate the protective role of DHA in insulin-secreting cells, MIN6 cells were treated with 40 μM DHA with or without 500 μM PA and then analyzed apoptosis by measuring caspase-3 cleavage and terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) assay. PA-induced apoptosis was defined as a remarkable increase in cleaved caspase-3 and TUNEL-positive red dots. Moreover, cotreatment with DHA for 24 hours significantly attenuated the level of cleaved caspase-3 and number of TUNEL-positive signals (Fig. 1), suggesting that DHA protects MIN6 cells against PA-induced apoptosis.
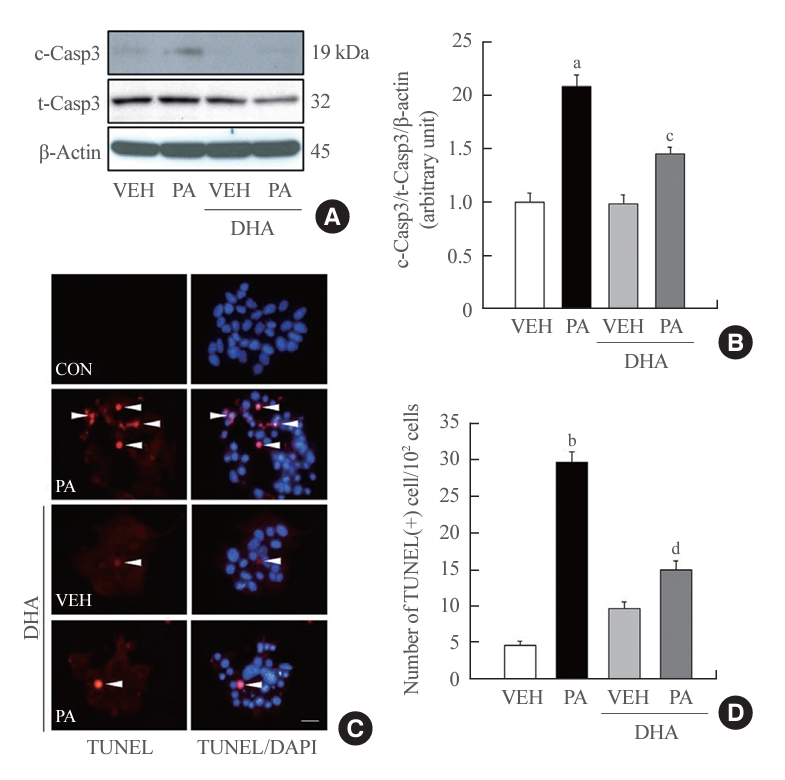
Docosahexaenoic acid (DHA) prevents palmitic acid (PA)induced apoptosis in insulin-secreting cells. (A, B) Mouse insulinoma 6 (MIN6) cells were incubated with 40 μM DHA in the presence or absence of 500 μM PA for 24 hours. Whole cell lysates were extracted and cleaved caspase-3 (c-Casp3) expression, an apoptosis marker, was measured by immunoblotting analysis. The ratio to total caspase-3 (t-Casp3)/β-actin was described (n=3). (C, D) After treatment with DHA and/or PA, MIN6 cells were fixed for terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) staining assay to observe apoptosis. Apoptotic cells were visualized with red dot signals (white arrows). The nuclei were indicated by 4’,6-diamidino2-phenylindole (DAPI). Scale bars=20 µm. The number of TUNEL-positive cells was counted and described as a ratio to 102 cells (n=3–4). VEH, vehicle; CON, control. aP<0.01, bP<0.001 compared with the VEH group; cP<0.05, dP<0.01 compared with the PA group.
DHA facilitates autophagy in MIN6 cells
Autophagy is a recycling process involving the encasing and dismantling of useless cellular organelles by autophagosomes [26]. Among the many factors involved in autophagy, LC3 is essential for the formation of autophagosomes. During autophagosome formation, LC3-I is converted to LC3-II; therefore, LC3-II is a definite indicator of autophagy. We found that DHA treatment obviously increased LC3-II conversion in MIN6 cells. In cells exposed to 30 or 40 μM DHA, the level of LC3-II protein was remarkably increased (Fig. 2A), and the distribution and intensity of the fluorescence signal indicated that LC3 proteins were greatly enhanced in 40 μM DHA-treated MIN6 cells (Fig. 2B). We also measured the autophagy-related factors p62 and Atg5. In the early phase of autophagy, p62 binds to ubiquitinated proteins and carries them to the autophagosome for proteasomal degradation. Then, p62 is degraded in the autophagosomes in the late phase. Therefore, p62 expression decreases through autophagy activation [27]. p62 protein levels were significantly reduced in MIN6 cells treated with 30 and 40 μM DHA (Fig. 2A), indicating autophagy activation by DHA. Atg5, another autophagy-related factor, induces LC3 lipidation and facilitates LC3-I conversion to LC3-II [28]. Atg5 protein levels were also markedly increased after DHA treatment (Fig. 2A).

Docosahexaenoic acid (DHA) upregulates autophagy dose-dependently in insulin-secreting cells. (A) Mouse insulinoma 6 (MIN6) cells exposed to DHA at 0, 10, 20, 30, and 40 μM for 24 hours. Expression of autophagy-related factors including light chain 3 (LC3) I/II, p62, autophagy-related 5 (Atg5) was measured by immunoblotting analysis. (B) MIN6 cells were treated with 40 μM DHA or vehicle for 24 hours, and analyzed by immunofluorescence to morphologically observe the LC3 expression level. LC3 was detected with green fluorescence (white arrowheads), and nuclei were indicated by 4’,6-diamidino2-phenylindole (DAPI). Scale bars=20 µm.
Autophagy is upregulated by PA, and DHA enhances autophagy in PA-challenged MIN6 cells
In several studies, autophagy activation by PA in pancreatic β-cells has been reported [29,30] and was also confirmed by our data (Figs. 3, 4). p62 and Atg5 protein levels were notably affected by PA treatment, and LC3-II levels were slightly increased (Fig. 3A, B). Despite autophagy activation by PA, DHA distinctly induced additional upregulation of autophagy compared with that of cells treated only with PA, showing a remarkable increase in LC3-II and Atg5 levels, with decreased p62 levels (Fig. 3A, B). We also observed localization of LC3 and Atg5 expression using an immunofluorescence assay. The fluorescence signal intensities for LC3 (green) and Atg5 (red) were greatly enhanced in DHA-treated cells, consistent with the increased protein levels in the immunoblotting assay. Double-positive cells that simultaneously expressed both LC3 and Atg5 were observed in the DHA-treated groups, and co-localization of LC3 and Atg5 was detected in DHA- and palmitate-treated cells (Fig. 3C). These data suggest that DHA enhances autophagy in insulin-secreting cells regardless of PA.
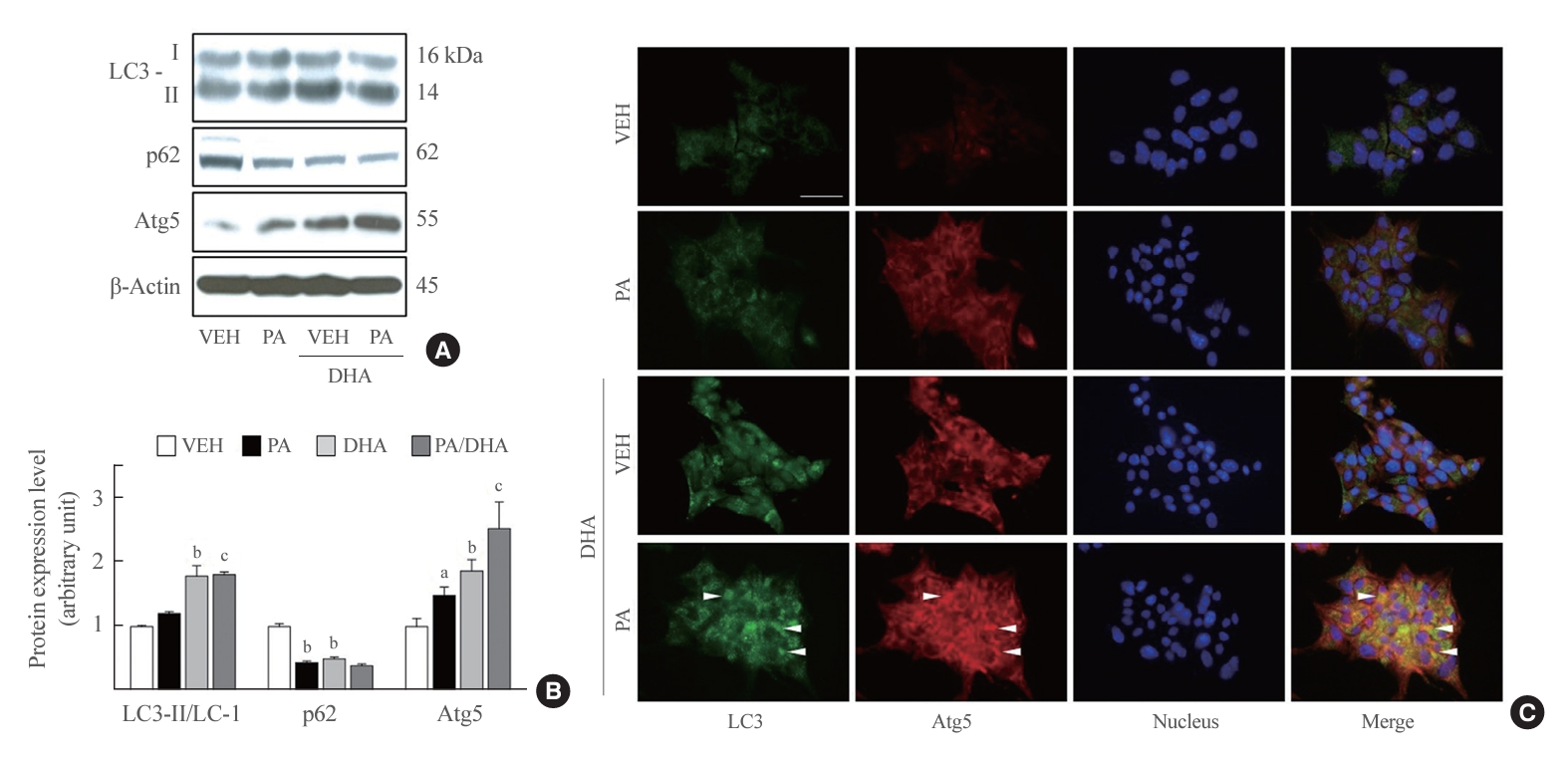
Docosahexaenoic acid (DHA) upregulates autophagy in a palmitate-treated manner in mouse insulinoma 6 (MIN6) cells. (A, B) Light chain 3 (LC3) I, II, p62, and autophagy-related 5 (Atg5) expression were measured in MIN6 cells after incubation with and without 500 µM palmitate and/or 40 µM DHA for 24 hours. The ratio to β-actin was identified (n=3–4). (C) LC3 and Atg5 expression was detected using double immunofluorescence. LC3 and Atg5 are respectively visualized by green and red fluorescence, and LC3 and Atg5 co-localization is indicated with white arrowheads. Nuclei were marked by 4’,6-diamidino2-phenylindole (DAPI). Scale bar=20 μm. VEH, vehicle; PA, palmitic acid. aP<0.05, bP<0.01 compared with the VEH group; cP<0.01 compared with the PA group.
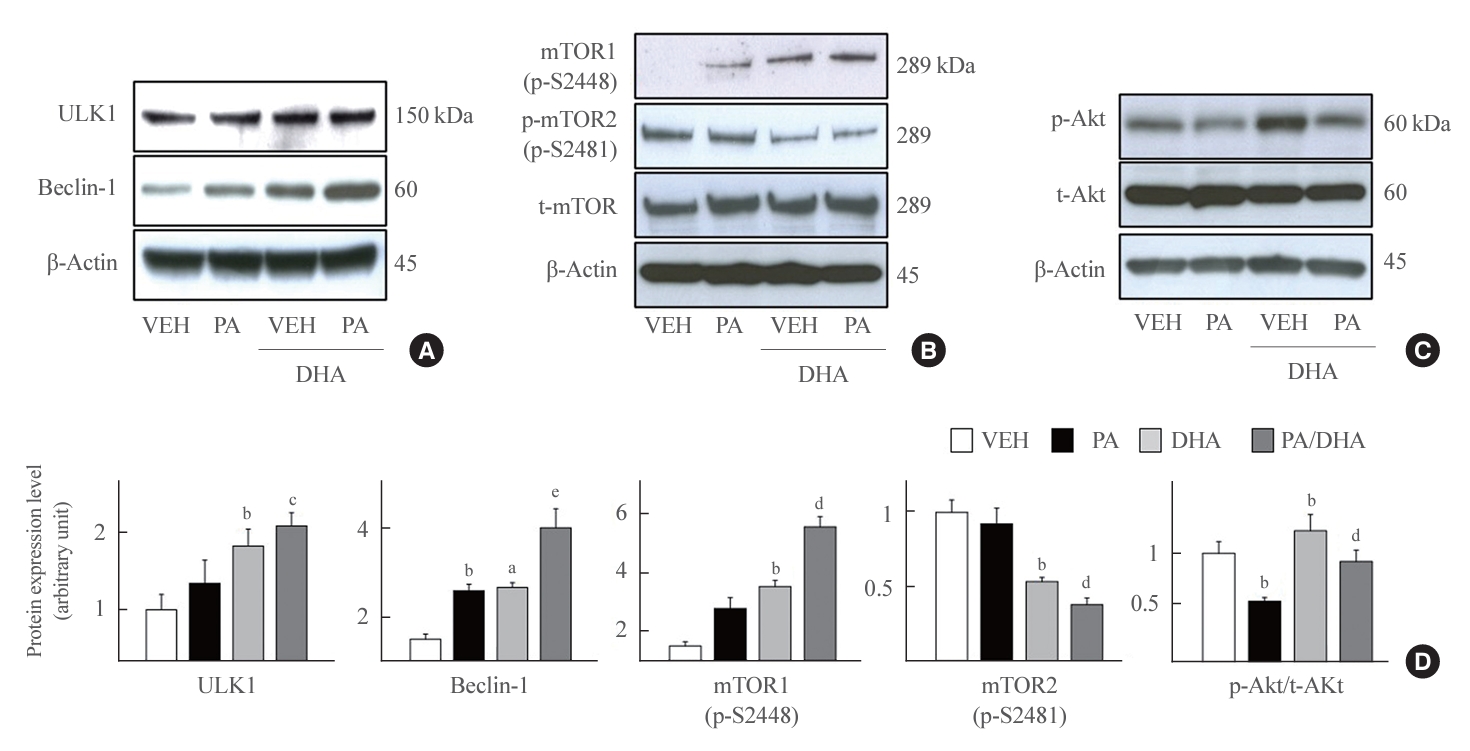
Docosahexaenoic acid (DHA) affects protein kinase B (Akt)/mammalian target of rapamycin (mTOR)/unc51 like autophagy activating kinase 1 (ULK1) axis. (A, B, C) Mouse insulinoma 6 (MIN6) cells were incubated with 40 μM DHA in the presence or absence of 500 μM palmitate for 24 hours. Whole cell lysates were extracted and analyzed by immunoblotting with antibodies specific for ULK1, Beclin-1, mTOR phosphorylated on S2448 or S2481, or total mTOR (t-mTOR), phosphorylated Akt (p-Akt), total Akt (t-Akt), and β-actin. (D) The ratio to β-actin was described. All values are expressed as the mean±standard error of the mean (n=3–4). VEH, vehicle; PA, palmitic acid. aP<0.05, bP<0.01 compared with the VEH group; cP<0.05, dP<0.01, eP<0.001 compared with the PA group.
DHA-induced autophagy is mediated by mTOR regulation
Autophagy is mainly induced in response to nutrient deprivation and is regulated by mTOR. The kinase mTOR is an essential controller of the autophagic process that is simultaneously promoted by starvation and cellular stressors [31]. Under starvation conditions, mTOR enhances ULK1 [32] and Beclin-1 expression [33] to initiate autophagosome formation. Therefore, we examined alterations in upstream factors of autophagy, such as ULK1, Beclin-1, and mTOR, to investigate cellular mediators of DHA-induced autophagy activation. DHA treatment consistently increased ULK1 and Beclin-1 expression in vehicle- or PA-treated conditions (Fig. 4A), although interestingly, the two phosphorylated forms of mTOR were contrarily affected by DHA. mTOR1 (p-S2448) levels increased after DHA treatment, whereas mTOR2 (p-S2481) levels significantly decreased after DHA treatment (Fig. 4B). Akt phosphorylation, which is known to be upstream of mTOR, also significantly increased after DHA treatment (Fig. 4C). These data indicate that the Akt/mTOR1/ULK1/Beclin-1 axis is a pivotal mediator of DHA-induced autophagy activation and that mTOR1 (p-S2448) is a key regulator of autophagy.
GPR120 is essential to induce autophagy activation by DHA
Next, we investigated whether GRP120 is involved in DHA-induced autophagy activation. GPR120, a cellular receptor for long-chain fatty acids, is expressed in pancreatic β-cells and stimulated by PUFAs [17]. Therefore, we estimated that GPR120 inhibition would be altered using AH7614, a chemical antagonist of GPR120, on the protein level, specifically Akt, mTOR, ULK1, Beclin-1, and LC3. GPR120 antagonism was obviously reversed by DHA-induced enhancement of p-Akt, p-mTOR1, ULK1, Beclin-1, and LC3-II (Fig. 5A). Moreover, blocking GPR120 using AH7614 alleviated the DHA-induced reduction in caspase-3 cleavage (Fig. 5B). These data suggest that GPR120 mediates DHA-induced autophagy activation and protects pancreatic β-cells against PA-induced apoptosis. The role of GPR120 was confirmed using GSK137647, a selective agonist of GPR120. GSK137647 treatment significantly enhanced p-Akt, p-mTOR1, ULK1, Beclin-1, and LC3-II levels (Fig. 6), suggesting that GPR120 stimulation by DHA upregulates autophagy mediated by the Akt/mTOR/ULK1/Beclin-1 axis in pancreatic β-cells.

Inhibition of G protein-coupled receptor 120 (GPR120) strictly blocks the docosahexaenoic acid (DHA) effect in mouse insulinoma 6 (MIN6) cells. MIN6 cells were pretreated with 20 μM AH7614 or dimethyl sulfoxide (DMSO) for 2 hours, and then incubated with palmitate/DHA for 24 hours. (A) Whole cell lysates (WCLs) were extracted and analyzed by immunoblotting with antibodies specific for phosphorylated protein kinase B (p-Akt), total Akt (t-Akt), mammalian target of rapamycin (mTOR) phosphorylated on S2448 or S2481, or total mTOR (t-mTOR), unc51 like autophagy activating kinase 1 (ULK1), Beclin-1 light chain 3 (LC3) II/I, and β-actin. (B) The ratio of each factor’s band intensity to β-actin was described. (C) Cleaved caspase-3 (c-Casp3) and total caspase-3 (t-Casp3) were measured by immunoblotting analysis, and the level of c-Casp3 was normalized to t-Casp3/β-actin. All values are expressed as the mean±standard error of the mean (n=3). VEH (V), vehicle; PA (P), palmitic acid; NS, non-significant. aP<0.01 compared with the VEH group of DMSO set; bP<0.05 compared with the PA group of DMSO set; cP<0.01 compared with VEH group of AH7614 set.

Autophagy activation by G protein-coupled receptor 120 (GPR120) agonist via protein kinase B (Akt)/mammalian target of rapamycin (mTOR) axis. Mouse insulinoma 6 (MIN6) cells were incubated with or without 500 μM palmitate and/or 50 μM GSK137647 for 24 hours. (A, B) Whole cell lysates were extracted and analyzed by immunoblotting with antibodies specific for phosphorylated Akt (p-Akt), total Akt (t-Akt), mTOR phosphorylated on S2448 or S2481, or total mTOR (t-mTOR), unc51 like autophagy activating kinase 1 (ULK1), Beclin-1 light chain 3 (LC3) I/II, and β-actin. All values are expressed as the mean±standard error of the mean (n=3–4). VEH, vehicle; PA, palmitic acid. aP<0.05, bP<0.001 compared with the VEH group; cP<0.05, dP<0.01, eP<0.001 compared with the PA group.
DISCUSSION
Saturated NEFAs have been a known cause of β-cell apoptosis and dysfunction in patients with diabetes and obesity for decades [34], and several studies have suggested the beneficial effects of PUFAs, such as EPA and DHA [35-37]. Moreover, a recent study reported that elongation of very long-chain fatty acids-like 2, an enzyme associated with de novo PUFA synthesis, is essential for protection against glucolipotoxicity-induced apoptosis [10]. However, the cellular mechanism underlying PUFA-induced β-cell protection remains unknown. In the present study, we showed that DHA induces autophagy activation and protects against PA-induced apoptosis, which is mediated by the GPR120/mTOR1 axis.
Recently, growing evidence has shown that autophagy activation is essential for maintaining homeostasis and viability in various cell types [38,39]. In particular, in pancreatic β-cells autophagy has been regarded as a protective mechanism against cellular stress through the removal of unusual organelles and toxic molecules [40,41]. Although several beneficial effects of PUFAs have been reported in pancreatic β-cells [10,17], autophagy activation by DHA was first suggested in this study.
Autophagy is induced by nutrient starvation and cellular stresses because mTOR is a key regulator of autophagy and the main sensor of cellular nutrient status and stress [42]. In our study, we clearly showed that DHA-induced autophagy activation is mediated by the mTOR signaling pathway, demonstrating the promotion of Akt, mTOR, ULK1, and Beclin-1 expression and increased LC3-II levels (Fig. 4). We discovered contrasting fluctuations in mTOR phosphorylation, depending on the phosphorylation site. Interestingly, phosphorylation on S2448 of mTOR was significantly increased, whereas phosphorylation of mTOR on S2481 was decreased after DHA treatment. mTORC, a protein complex, is classified into mTORC1 and mTORC2 according to binding molecules, raptor and rictor respectively, and mTORC1 inhibits autophagy [43]. Initially it was thought that S2448 and S2481 were phosphorylation residues specific for mTORC1 and mTORC2, respectively [43]. However, S2481 has been found to be phosphorylated in mTORC1 [44], and S2448 has been found to be phosphorylated in mTORC2 [45]. Therefore, we additionally measured raptor and rictor, indispensable markers of mTORCs, to presume the formation of mTORC1 and mTORC2. The amount of raptor protein was decreased by DHA, and rictor was not affected by DHA (Supplemental Fig. S1). Phosphorylation of S2448 residue was increased by DHA, whereas raptor was significantly reduced by DHA treatment. Indeed, reduction of raptor expression by DHA has already been reported in several studies [46,47]. Our results present the upregulation of phosphorylation on S2448 residue of mTOR, but do not suggest the enhancement of mTORC1. Further investigation is needed to reveal the role of phosphorylation on S2448 mTOR by DHA in autophagy regulation.
The beneficial role of GPR120 in pancreatic β-cells reportedly induces insulinotropic effects by regulating Akt and calcium (Ca2+) channels [48] and protects against lipotoxicity-induced β-cell dysfunction through the regulation of pancreatic and duodenal homeobox 1 (PDX1) expression and inhibition of inflammation [17]. However, the cellular mechanisms associated with β-cell improvement by GPR120 are still not fully understood. In this context, we demonstrated that a series of cellular signaling pathways, including specific phosphorylation on p-S2448 residue of mTOR, autophagy activation, and protection against apoptosis, are mediated by GPR120 stimulation by DHA. To date, autophagy induction by GPR120 stimulation has been observed in hepatocytes [49], bone marrow-derived stem cells [50], and macrophages [51], although not in pancreatic β-cells. We found that cell protection by GPR120 stimulation was mediated by mTOR phosphorylation and autophagy activation in pancreatic β-cells. We confirmed that GPR120 functions in the autophagy regulation and cell protection using AH7614 and GSK137647, specific GPR120 antagonists and agonists (Figs. 5, 6). Inhibition and stimulation of GPR120 by AH7614 and GSK137647 altered PA-induced mTOR phosphorylation, autophagy, and cellular apoptosis. These data suggest a potential role for GPR120 in the regulation of pancreatic β-cell autophagy and protection against lipotoxicity.
In diabetes-related toxic environments, including those with high glucose and NEFA levels, pancreatic β-cells are particularly exposed to heavy stresses because of their ability to produce and release insulin. Recently, regulation of the autophagy system has been considered to play a key role in overcoming stress [52]. In this context, we showed that DHA protects insulin-secreting cells against PA-induced apoptosis via autophagy activation and that this effect is mediated by GPR120 and specific mTOR phosphorylation on S2448. We suggest that DHA has therapeutic effects on damaged β-cells, and that the cellular signaling pathway promoted by DHA may be a new research target with potential pharmacotherapeutic implications for pancreatic β-cell protection.
Supplementary Material
Supplemental Fig. S1.
Docosahexaenoic acid (DHA) reduces raptor protein, but not affects rictor. Mouse insulinoma 6 (MIN6) cells were incubated with 40 μM DHA in the presence or absence of 500 μM palmitate for 24 hours. (A) Whole protein was extracted and analyzed by immunoblotting with antibodies specific for raptor, rictor, and β-actin. (B) The ratio to β-actin was described. All values are expressed as the mean±standard error of the mean (n=3). VEH, vehicle; PA, palmitic acid. aP<0.05 compared with the VEH group; bP<0.05 compared with the PA group.
Notes
CONFLICTS OF INTEREST
Won-Young Lee is an editor-in-chief and Eun-Jung Rhee is a deputy editor of the journal. However, they were not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
AUTHOR CONTRIBUTIONS
Conception or design: S.W.H., J.L., E.J.R., W.Y.L. Acquisition, analysis, or interpretation of data: S.W.H., J.L., S.J.M., H.K., S.E.P. Drafting the work or revising: S.W.H., J.L. E.J.R., W.Y.L. Final approval of the manuscript: S.W.H., J.L., S.J.M., H.K., S.E.P., E.J.R., W.Y.L.
Acknowledgements
This study was supported in part by a grant from the National Research Foundation (NRF), which is funded by the Korean government (NRF-2021R1F1A1057306), and by a grant from the Yuhan Corporation. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.