Incretin and Pancreatic β-Cell Function in Patients with Type 2 Diabetes
Article information

Abstract
To maintain normal glucose homeostasis after a meal, it is essential to secrete an adequate amount of insulin from pancreatic β-cells. However, if pancreatic β-cells solely depended on the blood glucose level for insulin secretion, a surge in blood glucose levels would be inevitable after the ingestion of a large amount of carbohydrates. To avoid a deluge of glucose in the bloodstream after a large carbohydrate- rich meal, enteroendocrine cells detect the amount of nutrient absorption from the gut lumen and secrete incretin hormones at scale. Since insulin secretion in response to incretin hormones occurs only in a hyperglycemic milieu, pancreatic β-cells can secrete a “Goldilocks” amount of insulin (i.e., not too much and not too little) to keep the blood glucose level in the normal range. In this regard, pancreatic β-cell sensitivity to glucose and incretin hormones is crucial for maintaining normal glucose homeostasis. In this Namgok lecture 2022, we review the effects of current anti-diabetic medications on pancreatic β-cell sensitivity to glucose and incretin hormones.
INTRODUCTION
Blood glucose levels are tightly regulated within a very narrow range in individuals with normal glucose tolerance, which is maintained by the coordinated action of islet and incretin hormones, as well as neural and humoral factors from different tissues and organs. Among them, the action of incretin hormones, known as the incretin effect, contributes to 50% to 70% of insulin secretion after the oral ingestion of glucose in healthy subjects [1]. To date, two incretin hormones have been discovered: glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) [2]. The amount of GLP-1 and GIP secretion is proportional to the amount of ingested carbohydrates [1,3], and these hormones send signals about the anticipated influx of glucose from the gut lumen so that the pancreatic β-cells secrete an appropriate amount of insulin to prevent excessive excursions of blood glucose levels. Both hormones augment insulin secretion in a glucose-dependent manner [4], which is a fail-safe mechanism that minimizes the risk of hypoglycemia even in a situation with excessive secretion of incretin hormones. Various pharmaceutical agents based on incretin hormones have been developed for the treatment of type 2 diabetes [5,6]. Considering the unique nature of glucose-dependent insulin secretion in response to incretin hormones, it would be timely to review the pancreatic β-cell response to glucose and incretin hormones and how it is modulated by current anti-diabetic medications.
IN VIVO ASSESSMENT OF PANCREATIC β-CELL SENSITIVITY TO BLOOD GLUCOSE AND INCRETIN HORMONES
Increased blood glucose levels and incretin hormones are the major stimuli of insulin secretion. Thus, in a simplified model as shown in the following equation, the insulin secretion rate is determined by β-cell glucose sensitivity and the blood glucose level, β-cell GLP-1 sensitivity and the blood GLP-1 level, and β-cell GIP sensitivity and the blood GIP level [7].
I, insulin; k1, β-cell glucose sensitivity; BG, blood glucose level; kglp-1, β-cell GLP-1 sensitivity; GLP-1,blood GLP-1 level; kgip, β-cell GIP sensitivity; GIP, blood GIP level
In vivo measurements of β-cell glucose sensitivity are made by calculating the slope of the insulin secretion rate versus the blood glucose level during a graded glucose infusion study, which gradually raises blood glucose levels, typically from 5 to 12 mmol/L (Fig. 1A) [8]. As illustrated in Fig. 1B, β-cell glucose sensitivity is markedly lower in patients with type 2 diabetes than in individuals with normal glucose tolerance [8].

In vivo assessment of β-cell glucose sensitivity using a graded insulin infusion protocol. (A) Estimation of β-cell glucose sensitivity by calculating the slope of the insulin secretion rate versus the blood glucose level. (B) Results showing that a single injection of liraglutide restored β-cell glucose sensitivity in patients with type 2 diabetes. Open triangles represent healthy control subjects. Open and closed rectangles represent type 2 diabetes subjects who received placebo or liraglutide, respectively. Adapted from Chang et al. [8], with permission from the American Diabetes Association. ISR, insulin secretion rate.
Meanwhile, β-cell incretin sensitivity is measured in vivo by performing a hyperglycemic clamp study with GLP-1 and/or GIP infusion (Fig. 2). The increment of insulin secretion in response to GLP-1 or GIP infusion during the hyperglycemic clamp study indicates the β-cell incretin sensitivity [9-11]. In one study, the infusion of GLP-1 and GIP increased plasma insulin levels by 4.1 and 3.6 times, respectively, compared to a saline infusion in healthy subjects during a hyperglycemic clamp procedure [10]. In the same study, β-cell incretin sensitivity was markedly lower in patients with type 2 diabetes [10].
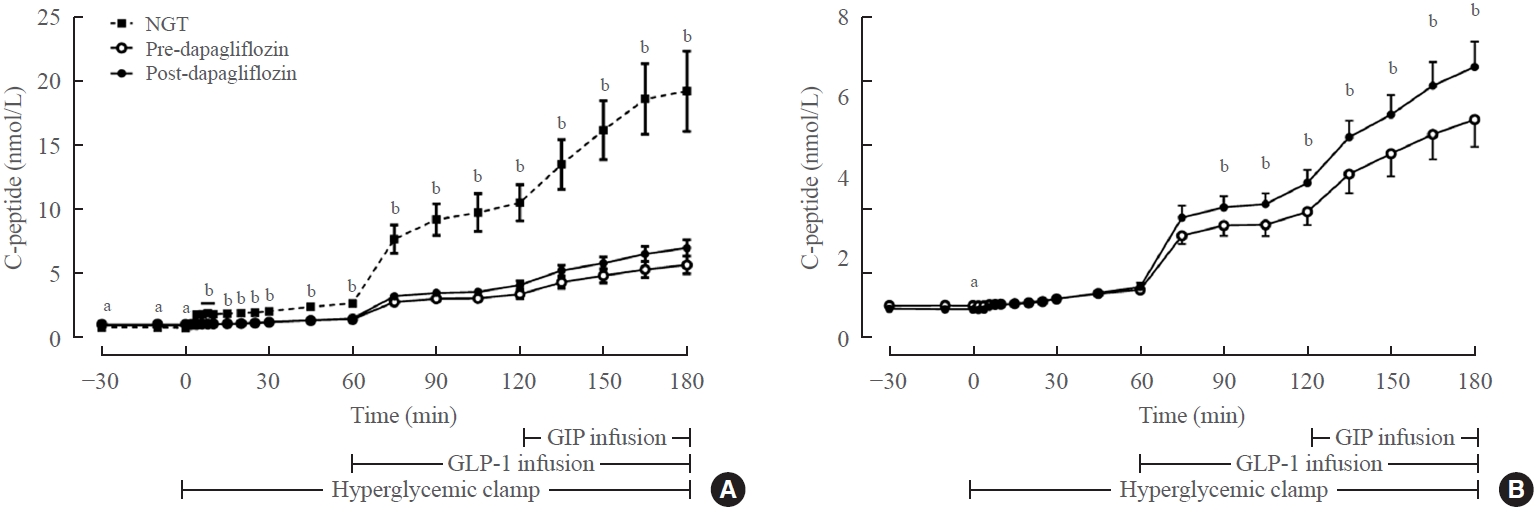
In vivo assessment of β-cell incretin sensitivity using a hyperglycemic clamp with glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) infusion. (A) C-peptide responses to hyperglycemia, GLP-1 infusion, and GIP infusion in subjects with normal glucose tolerance and type 2 diabetes. (B) C-peptide response to hyperglycemia, GLP-1 infusion, and GIP infusion in subjects with type 2 diabetes before and after dapagliflozin treatment (magnified from Fig. 2A). Adapted from Ahn et al. NGT, normal glucose tolerance. aP<0.05 for comparison between NGT and predapagliflozin studies; bP<0.05 for comparison between NGT and both pre- and postdapagliflozin studies.
INCRETIN THERAPY AND β-CELL GLUCOSE SENSITIVITY
Defective insulin secretion is the cornerstone of the pathophysiology of type 2 diabetes. As shown in Equation 1, pancreatic β-cell sensitivity to glucose, incretin, or both is critical for insulin secretion. Therefore, restoring pancreatic β-cell sensitivity to glucose and/or incretin hormones is essential for improving glycemic control in patients with type 2 diabetes. Since chronic hyperglycemia itself impairs β-cell function [12], pharmacologic interventions to reduce hyperglycemia may improve β-cell function and restore β-cell glucose sensitivity [13].
The effect of a GLP-1 receptor agonist, liraglutide, on β-cell glucose sensitivity was examined in a placebo-controlled crossover study involving 10 patients with well-controlled type 2 diabetes (mean hemoglobin A1c [HbA1c], 6.5%) and 10 healthy subjects [8]. Remarkably, a single injection of liraglutide restored β-cell glucose sensitivity, which became similar to that of healthy subjects [8]. In another study, 45 patients with poorly controlled type 2 diabetes (mean HbA1c, 8.4%) were randomized to canagliflozin, liraglutide, or both on top of their existing treatment regimen [14], and β-cell glucose sensitivity was calculated by a mathematical model using data from oral glucose tolerance tests with frequent sampling for insulin and C-peptide. Both canagliflozin and liraglutide significantly decreased HbA1c (by 0.89% and 1.43%, respectively) and increased β-cell glucose sensitivity (from 29±5 to 55±11 and from 33±6 to 101± 16, respectively). No meaningful additive effect was found in participants who received both canagliflozin and liraglutide. The magnitude of HbA1c reduction was closely correlated with the increase in β-cell glucose sensitivity, suggesting that alleviating glucose toxicity would be important for improving β-cell glucose sensitivity. However, it is noteworthy that incretin hormones may exert direct effects on pancreatic β-cell function by reducing endoplasmic reticulum stress, enhancing autophagy, and preventing apoptosis [15,16].
The effect of a dipeptidyl peptidase-4 (DPP-4) inhibitor on β-cell glucose sensitivity was calculated by mathematical modeling using data from a mixed meal test and a corresponding isoglycemic intravenous glucose infusion that exactly copied the blood glucose changes during the mixed meal test [17]. In that study, which involved 50 patients with type 2 diabetes (mean HbA1c, 7.4%), sitagliptin or placebo was given for 6 weeks. Compared to the placebo, sitagliptin significantly improved β-cell glucose sensitivity during the mixed meal test and the corresponding isoglycemic procedure, but β-cell glucose sensitivity failed to reach the level of control subjects without diabetes [17].
PHARMACOTHERAPIES TO IMPROVE β-CELL INCRETIN SENSITIVITY
Insulin secretion from pancreatic β-cells in response to incretin hormones is determined by the tissue levels of incretin hormones and signal transduction through incretin receptors. Although there have been mixed results with regard to the amount of incretin secretion, a meta-analysis showed that the secretion of GLP-1 and GIP is not reduced in patients with type 2 diabetes compared to their counterparts without diabetes [18,19]. Therefore, the secretion of GLP-1 and GIP does not play a critical role in impaired insulin secretion in patients with type 2 diabetes in general. However, in special situations with exposure to a massive amount of GLP-1 receptor agonists, such as in GLP-1 analog treatment or post-gastric bypass surgery, the tissue level of GLP-1 or its analog is important for determining pancreatic β-cell function. Otherwise, the expression level of the receptors for GLP-1 and/or GIP (hereafter, GLP-1R and GIPR, respectively) in pancreatic β-cells is a critical factor in determining the action of the incretin hormones [20].
Both genetic and environmental factors contribute to the expression level of GLP-1R and GIPR. Decreased GLP-1R and GIPR expression was observed in islets isolated from carriers of the type 2 diabetes risk allele of transcription factor 7 like 2 (TCF7L2) (rs7903146) [21]. In addition, individuals harboring the type 2 diabetes risk allele of Wolfram syndrome 1 gene (WFS1) (rs10010131) displayed reduced β-cell GLP-1 sensitivity as assessed by a hyperglycemic clamp with GLP-1 infusion [22]. It is also noteworthy that hyperglycemia itself may reduce incretin hormone receptor expression by mechanisms including glucose toxicity [23]. Several medications have the potential to restore β-cell sensitivity to incretin hormones (Table 1). In addition, downstream of the GLP-1 receptor, β-arrestin is an important modulator of the action of GLP-1 in pancreatic β-cells by regulating the internalization of GLP-1R [24]. A recent large genome-wide association study of type 2 diabetes patients who were treated with GLP-1R agonists reported that low-frequency variants of ARRB1 encoding β-arrestin were associated with better responses to GLP-1R agonists [25]. These variants may explain the pharmacogenomic differences in the efficacy of GLP-1R agonists among different ethnicities.

Effects of Pharmacotherapies on the β-Cell Incretin Sensitivity
Insulin
In the current armamentarium for the treatment of type 2 diabetes, insulin is the most effective drug to rapidly correct hyperglycemia. In a small study including eight obese patients with poorly controlled type 2 diabetes, intensive insulin therapy was provided for 4 weeks, targeting a pre-meal blood glucose level of 5 mmol/L [10]. After the treatment, HbA1c decreased from 8.6% to 7.4%. A hyperglycemic clamp with GIP and GLP-1 infusion was performed before and after intensive insulin treatment. Insulin secretion in response to the GLP-1 and GIP infusion increased by 3.4- and 5.6-fold from the baseline values after intensive insulin therapy. The basal insulin secretion without GLP-1 and GIP infusion also increased, suggesting that intensive insulin therapy improved β-cell function in general and β-cell incretin sensitivity in particular. Since chronic hyperglycemia imposes deleterious effects on β-cell function by building up glucose toxicity [26], ameliorating glucose toxicity with intensive insulin therapy may restore β-cell function [27,28] and improve β-cell incretin sensitivity through a similar mechanism.
Sulfonylureas
Sulfonylureas stimulate insulin secretion in a non-glucose-dependent manner by closing the ATP-sensitive potassium channel (KATP) channel. These medications potentiate β-cell responses not only to glucose but also to non-glucose stimuli [29]. A small Canadian study administered glyburide for a month to five newly diagnosed type 2 diabetes patients, and glyburide decreased their fasting blood glucose levels from 12.8±1.8 to 8.5±0.8 mmol/L [9]. A hyperglycemic clamp with GIP infusion performed before and after glyburide treatment showed improvements in β-cell glucose sensitivity by 58% and β-cell GIP sensitivity by 130% [9]. Like insulin, sulfonylureas also improve both β-cell glucose sensitivity and GIP sensitivity by ameliorating hyperglycemia in patients with poorly controlled type 2 diabetes.
Metformin
The mechanism of action of metformin in glycemic control is very complex and involves multiple pathways. With regard to the incretin system, metformin is both an enhancer of GLP-1 secretion and a sensitizer of GLP-1 and GIP action [30]. Metformin stimulates GLP-1 secretion from the intestine and increases plasma levels of GLP-1 both in mice and humans [31,32]. The underlying mechanism of GLP-1 secretion in response to metformin is poorly understood. However, a possibility is that metformin may inhibit the apical sodium-dependent bile acid transporter, which in turn increases the concentration of bile acids in the intestinal lumen [33]. Then, the increased bile acids in the lumen may activate the G-protein-coupled receptor, Takeda-G-protein receptor 5 (TGR5) signaling, which stimulates GLP-1 secretion from enteroendocrine L-cells [34]. In addition, metformin may directly stimulate GLP-1 secretion by modulating AMP-activated kinase (AMPK) activity based on an ex vivo experiment using human ileal and colonic tissue [35]. Of note, metformin and a DPP-4 inhibitor additively increase active GLP-1 levels in the plasma [32], which confers clinical benefits in the management of type 2 diabetes.
Intriguingly, some clues suggest a GLP-1 sensitizing action of metformin in pancreatic β-cells. It was reported that metformin directly increased GLP-1R expression in the INS-1 β-cell line [31]. Metformin also increased both GLP-1R and GIPR levels in mice in a peroxisome proliferator-activated receptor α (PPAR-α)-dependent manner [31]. In our own study, 4-week metformin treatment also increased GLP-1R expression in pancreatic β-cells in Goto-Kakizaki rats, a non-obese type 2 diabetes animal model [36]. However, we did not observe any increase in GLP-1R expression after fenofibrate, a PPAR-α agonist, treatment. The discrepancy between these two studies might be explained by the nature of endogenous or pharmacologic ligands for PPAR-α and different animal species and experimental settings.
DPP-4 inhibitors
The main action of DPP-4 inhibitors is augmenting active GLP-1 and GIP levels in the plasma by inhibiting their enzymatic degradation. In addition to this, DPP-4 inhibitors may increase the action of incretin hormones. In a study involving 24 patients with inadequately controlled type 2 diabetes using metformin at baseline (mean HbA1c, 7.8%), sitagliptin or placebo was given on top of metformin for 12 weeks [37]. The GIP or GLP-1 sensitivity of β-cells was estimated by performing a hyperglycemic clamp with the infusion of saline, GIP, or GLP-1. The baseline GLP-1 sensitivity was 3- to 4-fold higher than that of GIP. Interestingly, sitagliptin treatment improved only β-cell GIP sensitivity by 35% compared to the placebo [37]. It was not clear why β-cell GLP-1 sensitivity did not improve in response to sitagliptin in that study. However, the baseline GLP-1 sensitivity was not exactly evaluated because the baseline hyperglycemic clamp was performed 2 to 7 days after treatment initiation [37].
Sodium-glucose co-transporter 2 inhibitors
Phlorizin (a non-specific sodium-glucose co-transporter [SGLT] 1/SGLT2 inhibitor) increased both GLP-1R and GIPR expression in the pancreatic islets of 90% pancreatectomized hyperglycemic rats [38]. Zucker diabetic fatty rats showed decreased GIPR expression in the pancreatic islets, which was restored by phlorizin treatment [39]. A pancreatic perfusion study revealed that insulin secretion improved during GIP infusion in phlorizin-treated rats as compared with vehicle-treated rats [39]. The effects of phlorizin on the incretin sensitivity of pancreatic β-cells suggest that selective SGLT2 inhibitors may also have such salutary effects.
We previously measured the incretin sensitivity of pancreatic β-cells in 10 subjects with normal glucose tolerance and 19 subjects with type 2 diabetes using a hyperglycemic clamp with the sequential addition of GLP-1 and GIP infusions [11]. Eight-week dapagliflozin treatment in patients with type 2 diabetes reduced the mean HbA1c from 7.9% to 7.1%. Dapagliflozin treatment significantly increased insulin secretion upon sequential addition of GLP-1 and GIP during the hyperglycemic clamp study (Fig. 2). The improvements in β-cell sensitivity to glucose, GLP-1, and GIP were closely correlated with the improvement of HbA1c, suggesting that the amelioration of glucose toxicity could be an important factor for improving pancreatic β-cell sensitivity to glucose, GLP-1, and GIP in patients with type 2 diabetes.
CLINICAL IMPLICATIONS OF β-CELL SENSITIVITY TO INCRETIN HORMONES
As discussed above, metformin has the unique property of being both an incretin enhancer and an incretin sensitizer. Therefore, a combination of metformin and incretin-based drugs would be a good therapeutic option for the treatment of type 2 diabetes. Indeed, the combination of a GLP-1R agonist or DPP-4 inhibitor with metformin provided additive glycemic improvements in patients with inadequately controlled type 2 diabetes [40-42].
Combining incretin-based drugs and sulfonylureas is another commonly used treatment option for type 2 diabetes. Although DPP-4 inhibitors are known to have a very low risk for hypoglycemia based on their mechanism of action, the concomitant use of a sulfonylurea increases the hypoglycemia risk [43]. In Japan, during the first 6 months after the launch of sitagliptin, cases of severe hypoglycemia increased when sitagliptin was added onto a sulfonylurea, especially in elderly patients with renal insufficiency [44]. Interestingly, there is cross-talk between sulfonylurea and incretin signaling involving the activation of the exchange protein activated by cyclic-AMP 2A (Epac2A)-Ras-like small GTPase 1 (RAP1) pathway, which may augment insulin secretion from pancreatic β-cells upon exposure to the combination of both drugs [45,46]. Therefore, caution should be exercised to prevent hypoglycemia when adding incretin-based therapy to pre-existing sulfonylurea therapy in patients with type 2 diabetes.
Regarding the positive effect of SGLT2 inhibitors on β-cell sensitivity to incretin hormones, the combination of incretin-based therapies and SGLT2 inhibitors could be a very effective option for anti-diabetic treatment. In our systematic review and meta-analysis evaluating the clinical efficacy and safety of combinations of DPP-4 inhibitors and SGLT2 inhibitors in patients with inadequately controlled type 2 diabetes, the combination of an SGLT2 inhibitor and a DPP-4 inhibitor showed a greater reduction in HbA1c (weighted mean difference, −0.6%), fasting plasma glucose, and 2-hour postprandial plasma glucose levels than the combination of a placebo with a DPP-4 inhibitor [47]. However, unlike our expectations, a synergistic effect of SGLT2 inhibitor and DPP-4 inhibitor use was not observed in these clinical trials, which could be attributed, at least partially, to the opposite action on glucagon secretion by the two classes of drugs.
CONCLUSIONS
For normal postprandial glucose metabolism, it is crucial to maintain adequate β-cell glucose, GLP-1, and GIP sensitivity. Several anti-diabetic medications have shown the potential to improve β-cell incretin sensitivity (Table 1). Evidence indicates that sulfonylureas, DPP-4 inhibitors, SGLT2 inhibitors, and insulin improve β-cell incretin sensitivity in humans. In addition, experimental evidence for metformin and phlorizin has been reported in animal studies. The improvement of β-cell incretin sensitivity by these different classes of anti-diabetic medications suggests a shared mechanism: the amelioration of glucose toxicity (Fig. 3), which provides a mechanistic insight into the use of combination treatment including incretin-based therapy for type 2 diabetes.
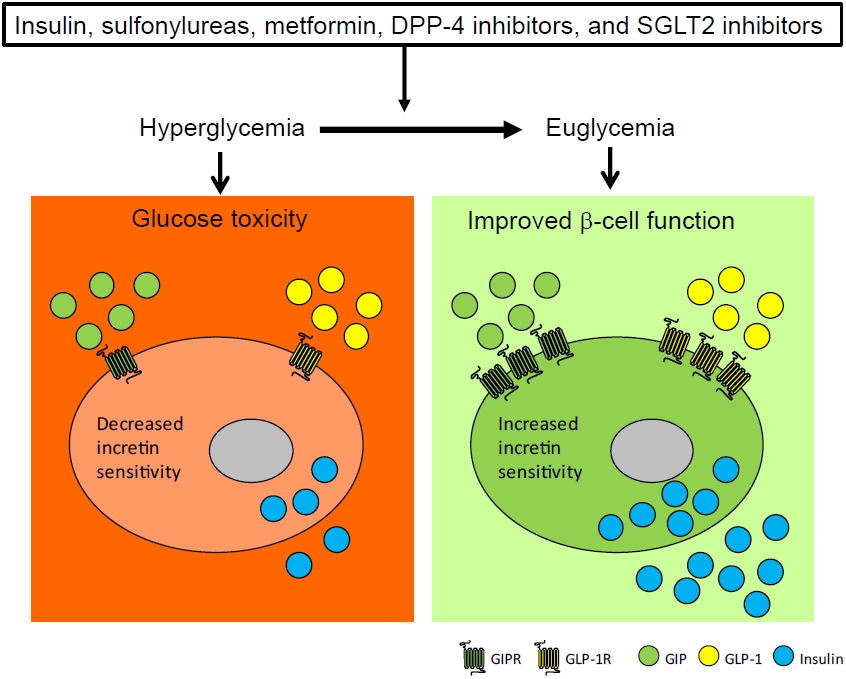
A proposed mechanism explaining how anti-diabetic medications improve β-cell incretin sensitivity. Anti-diabetic medications including insulin, sulfonylurea, metformin, dipeptidyl peptidase-4 (DPP-4) inhibitor and sodium-glucose co-transporter 2 (SGLT2) inhibitor restores pancreatic β-cell glucose and incretin sensitivity by ameliorating hyperglycemia. GIPR, glucose-dependent insulinotropic polypeptide receptor; GLP-1R, glucagon-like peptide-1 receptor; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1.
Notes
The Namgok Award is the highest scientific award of the Korean Endocrine Society, and is given to honor an individual who has made excellent contributions to progress in the field of endocrinology and metabolism. The Namgok Award is named after the pen name of Professor Hun Ki Min, who founded the Korean Endocrine Society in 1982. Professor Young Min Cho received the Namgok Award at the 10th Seoul International Congress of Endocrinology and Metabolism of the Korean Endocrine Society in October 2022.
CONFLICTS OF INTEREST
Young Min Cho received research grants from Daewoong Pharmaceuticals and Sanofi, and consultation fees from LG Chemical.