Recent Progress in the Medical Therapy of Pituitary Tumors
Article information

Abstract
Management of pituitary tumors is multidisciplinary, with medical therapy playing an increasingly important role. With the exception of prolactin-secreting tumors, surgery is still considered the first-line treatment for the majority of pituitary adenomas. However, medical/pharmacological therapy plays an important role in controlling hormone-producing pituitary adenomas, especially for patients with acromegaly and Cushing disease (CD). In the case of non-functioning pituitary adenomas (NFAs), pharmacological therapy plays a minor role, the main objective of which is to reduce tumor growth, but this role requires further studies. For pituitary carcinomas and atypical adenomas, medical therapy, including chemotherapy, acts as an adjuvant to surgery and radiation therapy, which is often required to control these aggressive tumors. In the last decade, knowledge about the pathophysiological mechanisms of various pituitary adenomas has increased, thus novel medical therapies that target specific pathways implicated in tumor synthesis and hormonal over secretion are now available. Advancement in patient selection and determination of prognostic factors has also helped to individualize therapy for patients with pituitary tumors. Improvements in biochemical and “tumor mass” disease control can positively affect patient quality of life, comorbidities and overall survival. In this review, the medical armamentarium for treating CD, acromegaly, prolactinomas, NFA, and carcinomas/aggressive atypical adenomas will be presented. Pharmacological therapies, including doses, mode of administration, efficacy, adverse effects, and use in special circumstances are provided. Medical therapies currently under clinical investigation are also briefly discussed.
INTRODUCTION
Pituitary tumors, according to the World Health Organization, are classified as typical or atypical adenomas and carcinomas [1]. They are usually slow growing and induce clinical manifestations either by mass effect on surrounding brain structures, or by causing hormonal imbalances. Mass effect is usually managed surgically. Partial or panhypopituitarism can result from compression of normal pituitary tissue by a growing mass, and is usually managed by hormonal replacement. Pituitary hyperfunction involves a more complex interpretation of patient signs and symptoms and correlation with laboratory investigation. Careful examination of pituitary function should be pursued at diagnosis and serially during follow-up.
Except for prolactin (PRL)-secreting tumors, management of other pituitary tumors includes surgery as a first-line treatment; however, preoperative use of medical therapy in patients with acromegaly and Cushing disease (CD) is on the increase. Currently, in most countries, medical therapy is used as a first-line treatment for prolactinomas, and as a second-line treatment for other secreting pituitary tumors (either persistent or recurrent) or in tumors with an aggressive course where multiple modalities (including surgery and radiation) are required.
MEDICAL TREATMENT OF CUSHING DISEASE
Medical therapy to achieve control of hypercortisolism is required if surgery is contraindicated, non-curative, delayed, or if there is no visible pituitary tumor on imaging.
There are three pathophysiological mechanism targets: central inhibition of adrenocorticotropic hormone (ACTH) secretion, adrenal-directed inhibition of steroidogenesis, and glucocorticoid-receptor blockade (Table 1).

Medical Therapy of Cushing Disease
Central inhibition of adrenocorticotropic hormone secretion
Anti-secretory and antiproliferative properties of somatostatin receptors ligands (SRLs) act at the CD source; corticotroph adenoma. However, hypercortisolism suppresses somatostatin receptor (SSTR) type 2, and first generation SRLs targeting this receptor (octreotide and lanreotide) are futile as an initial therapy. Their use, alone or in combination, once eucortisolism is achieved may be an option, but more studies are needed [2].
Pasireotide is a SRL with 40-time higher affinity for SSTR type 5, the predominant SSTR expressed in corticotroph tumors. It has been shown to suppress ACTH secretion and cell proliferation. Pasireotide (600 to 900 µg subcutaneous twice daily) induced >50% reduction of urinary free cortisol (UFC) in half the patients studied and approximately 20% achieved normal UFC [34]. A treatment response can be observed within 2 months and to date there is only one case of long-term treatment escape [5]. Tumor volume reduction (TVR) >20% has been observed in patients with baseline visible tumor on magnetic resonance imaging (MRI) [4]. Side effects include gastrointestinal symptoms, cholelithiasis; of note, hyperglycemia occurs in two-thirds of patients with CD [4]. Pasireotide long-acting release (LAR) administered monthly has also been shown in phase III trials to be effective in approximately half of patients with mild CD; overall remission rates were lower, similar with the subcutaneous formulation and with comparable incidence of hyperglycemia [6].
Cabergoline is the most effective dopamine agonist (DA) either as monotherapy or add-on combination therapy. A response rate of approximately 30% to 40% at a high mean dose of 3.5 mg/week (0.5 to 7 mg/week), has been reported [78], but with a lack of long-term response in some patients and concerns about valvulopathy at high doses.
Promising new molecular targets have been identified, including retinoic acid receptors, cyclin-dependent kinases and epidermal growth factor receptor (EGFR) with several drugs in different stages of clinical or preclinical development.
Adrenal-directed inhibition of steroidogenesis
Ketoconazole, an imidazole, has been used off-label to treat Cushing syndrome for 30 years, specifically acting on the cholesterol side-chain cleavage complex (P450scc) and 17- hydroxylase/17,20-lyase activity, reducing glucocorticoid synthesis. Concomitant inhibition of androgen synthesis can result in hypogonadism and gynecomastia in men, but can be beneficial in women with hirsutism. Ketoconazole efficacy varies from 30% to 80%, with the controversial hypothesis of an additional central modulating effect since ACTH levels remain unchanged on therapy [91011]. Approximately 20% of ketoconazole treated patients will eventually escape [9] and some will experience tumor progression. Tolerance may be limited by nausea or vomiting, and hepatotoxicity has to be regularly monitored.
Metyrapone inhibits 11β- and 19-hydroxylase and has potent cortisol-lowering effects; approximately 50% of patients will achieve eucortisolism [12]. An initial compensatory increase in ACTH might attenuate clinical response during the first months of therapy. Side effects include hyperandrogenism in women, and deoxycorticosterone accumulation can result in edema, hypokalemia, and hypertension.
Mitotane is mainly used for its adrenolytic effects in adrenocortical cancer, but at lower doses also has a slow, but potent inhibitory effect on steroid biosynthesis. Approximately 70% of patients will achieve normal UFC and about 10% may have sustained remission after discontinuation [1314]. Adrenal insufficiency (AI) is frequent and higher doses of hydrocortisone are needed for replacement, due to mitotane-induced increase in corticosterone-binding globulin and CYP3A4 inducer effect increasing cortisol metabolism. Side effects include gastrointestinal symptoms, impaired mental functions, gynecomastia, dizziness, and hyperlipidemia; thus, limiting long-term use.
Etomidate, an anesthetic agent, is the only parenteral drug. A low, non-hypnotic perfusion dose, can normalize cortisol levels in patients acutely ill or preoperatively in CD patients with uncontrolled hypercortisolism [14]. Patients have to be monitored for altered consciousness in an intensive care unit.
Medical therapies currently in clinical trial include an oral inhibitor of 18-hydroxylase; osilodrostat (LCI699). In a multicenter 22-week open-label study, normalization of UFC was attained within 10 weeks in 78% of patients. Hypokalemia is reportedly the most frequent side effect [1516] and two phase III studies are ongoing [17]. Levoketoconazole, an enantiomere of ketoconazole, is also under phase III trial and based on preclinical data, may have better efficacy and an advantageous side effect profile compared to ketoconazole [17].
Glucocorticoid-receptor blockade
Mifepristone acts as a glucocorticoid-receptor antagonist, with >10-fold affinity for glucocorticoid-receptor compared to cortisol and >3 times compared to dexamethasone [18]. Mifepristone blocks the progesterone-receptor and is contraindicated in women planning pregnancy. Patients must be monitored closely for hypokalemia, edema, and worsening hypertension. ACTH is expected to increase in two-thirds of patients without tumor growth correlation, but patients with macroadenomas should be monitored for tumor progression [19]. Importantly, laboratory evaluation for cortisol status is not reliable and doses adjustments should be based on clinical status [1820]. If AI occurs, high doses of dexamethasone (2 to 10 mg daily) are often required to overcome receptor blockade [20].
Combination therapy is sometimes needed to achieve eucorti solism in patients with more severe CD or when tolerance to a single agent is limited [2122]. Steroidogenic inhibitors can be combined, the most frequent being ketoconazole and metyrapone, with good results. Cabergoline and ketoconazole was shown to normalize UFC in almost 80% of CD patients in one study, and add-on pasireotide can provide a 10% supplementary effect (Table 1) [1723].
MEDICAL TREATMENT OF ACROMEGALY
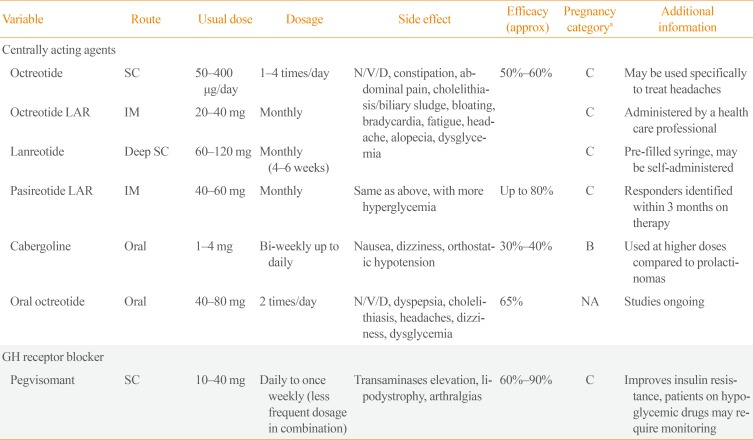
Medical therapy to control excess growth hormone (GH) secretion is usually reserved for patients with uncontrolled disease after an initial surgery or who are not surgical candidates. Therapeutic options include two classes of pharmacologic agents: central-acting inhibitors of GH secretion, including SRLs and DAs, and pegvisomant, a GH-receptor blocking agent (Table 2).

Medical Therapy of Acromegaly
Central inhibition of growth hormone secretion
For decades SRLs have been the first line medical treatment for acromegaly. First generation SRLs, octreotide LAR and lanreotide are considered equivalent medical therapies with minor differences in affinity for SSTR subtypes. Clinical action is mediated mainly via SSTR2A. These SRLs are effective in approximately 50% to 60% of patients in normalizing GH and insulin-like growth factor 1 (IGF-1; lower rates in non-selected or patients without surgery) [24], but also may act specifically on acromegaly-induced headache [25]. Significant TVR of >20% is also observed in ~65% of patients [2627] and shrinkage is more pronounced when SRLs are used as a first-line therapy [26]. Side effects include gastrointestinal symptoms, cholelithiasis/biliary sludge, hyper- or hypoglycemia, and bradycardia.
Preoperative SRL use may be considered in patients with severe pharyngeal thickness and sleep apnea syndrome to reduce perioperative morbidity [25]. Tumor shrinkage, without a clear improvement in surgical cure rate, has been shown, but evidence is lacking as to a precise role and duration of pre-surgical medical treatment [25].
Predictors of good response to first generation SRLs include densely granulated tumors [2829] (typically older patients and MRI T2-hypointensity signal [30]), lower Ki-67 expression and SSTR2A positive immunostaining [31]. Patients with familial acromegaly and aryl hydrocarbon receptor interacting protein (AIP) mutations tend to be resistant to first generation SRLs [32]. Rarely, disease remission has been described with first generation SRL treatment and withdrawal may be attempted in selected patients [3334]. Overall, 10% of patients are considered completely SRL-resistant and 40% to 50% will have an incomplete response, requiring alternative or combination therapies [2931].
Pasireotide, a second generation SRL, has been shown to normalize IGF-1 in 20% of patients resistant to first generation, with similar or slightly greater TVR [35] and may prove more beneficial in patients with sparsely-granulated tumors or with AIP mutations [36]. Worsening hyperglycemia, especially in patients with pre-existing diabetes, is observed in 50% of patients and close observation and treatment is needed [37].
Cabergoline can be effective alone or in combination by inhibiting GH secretion in patients with mild acromegaly (<1.5×upper limit of normal [ULN] elevation in IGF-1). Mixed GH-PRL tumors (PRL positive immunostaining) and hyperprolactinemia do not predict response [3839].
Growth hormone-receptor blockade
Pegvisomant is a GH-receptor antagonist, and blocks peripheral production of liver IGF-1 by competing with endogenous GH. Pegvisomant has a long half-life (60 to 138 hours) and can be used daily or with reduced frequency, up to once weekly. Dose-dependent normalization of IGF-1 levels was achieved in 60% of patients at mean dose of 18 mg/day, and up to 90% of cases at doses of up to 40 mg/day [4041]. Patients with insulin-treated diabetes tend to require higher doses because of a possible up-regulation of the hepatic GH-receptor by hyperinsulinemia [42]. Injection-site rotation is important as patients may rarely develop lipodystrophy (especially women), and liver function has to be regularly monitored. Mild transaminases elevation (2 to 5 times normal) is expected; however, if more severe (>5 times normal) dose reductions or discontinuation is required. By blocking IGF-1 production, GH will increase and thus cannot be monitored for disease-control. Approximately 3% to 5% of patients may experience tumor growth, and serial imaging is important in patients with residual tumors [40].
For patients needing combination therapy, first generation SRL and pegvisomant results in 80% to 97% normal IGF-1 at lower doses of pegvisomant, with better tolerability and at lower cost, but patients require close monitoring, notably for liver enzyme elevations [4344]. Also, the addition of cabergoline to an SRL, can have additional effect in achieving control in patients with mild (<1.5 times ULN) elevation in IGF-1 [43].
Medical therapies currently in clinical trial include a phase III study of an oral form of octreotide where a sustained response was noted in 85% of patients initially controlled on the injectable form, with similar tolerance [45]. Long-acting lanreotide (up to 3 months interval), subcutaneous long-acting octreotide, octreotide implants, somatoprim (SRL with additional SSTR type 4 affinity and less insulin inhibition), and antisense oligonucleotides, which downregulates GH-signaling, are also in different stages of clinical or preclinical development.
MEDICAL TREATMENT OF PROLACTINOMAS
Dopamine has an inhibitory effect on lactotroph cells acting through dopamine-receptor subtype 2 (D2). Bromocriptine and cabergoline (Table 3) act more specifically on this receptor subtype, and are therefore the cornerstone of therapy for PRL-secreting tumors. DAs have a dual action; inhibition of PRL secretion per se, and induction of apoptosis in lactotroph cells [46]. They have quick onset of action and usually TVR parallels the decrease in serum PRL, but response can differ depending on prolactinoma subtype. If there is erosion of the sella floor by a large macroprolactinoma, cerebrospinal fluid leak can be a consequence of rapid tumor shrinkage and patient awareness of new, persistent rhinorrhea is advised. Nausea and dizziness are usually transient and improve if medication is taken with food, before bedtime. An increase in compulsive disorders (e.g., hypersexuality and gambling) has been noted in 5% of patients on DAs [47]. Cabergoline is more potent with better tolerance profile than bromocriptine [46]; however, safety data in pregnancy is greater with bromocriptine. DAs are usually discontinued as soon as pregnancy is confirmed, but sometimes have to be reinstituted if there is significant tumor progression during pregnancy. Of note, for smaller prolactinomas without mass effect, replacement with testosterone or estrogen±progesterone can be a reasonable option for hypogonadism [46] with careful biochemical and structural monitoring since estrogen can stimulate tumor growth.

Medical Therapy of Prolactinomas
DAs resistance is defined as failure to normalize PRL levels or failure to achieve TVR of >50% at maximal usual doses of medication (bromocriptine 7.5 mg/day or cabergoline 2 mg/week). Treatment resistance can be due to poor compliance, concomitant estrogen or rarely testosterone replacement, or tumor transformation to a more atypical/aggressive type [48]. Structural and/or biochemical resistance can be as high as 25% on bromocriptine, and if so, switching to cabergoline may achieve further response in 70% to 80% of those patients. Increasing the cabergoline dose up to 4 to 6 mg/week in patients with objective response is a good approach; and some groups reported doses as high as 12 mg/week [49]. Higher doses may induce cardiac valvular abnormalities by a serotonin-mediated stimulatory effect on fibroblasts [48]. Recent prospective studies did not show a clear association with smaller doses [50], but should be monitored on cabergoline doses >2 mg/week [46]. Overall, 10% of patients will remain DA resistant and will require surgery, which can also be considered for patients intolerant to medication or with larger tumors pre-pregnancy.
Lapatinib, a tyrosine-kinase inhibitor targeting EGFR and erbB2 tyrosine-kinase, approved in the treatment of breast cancer, has been shown in animal models to inhibit PRL secretion, expression and EGFR/HER2 signaling [51]. Cooper et al. [52] reported a positive response in two patients resistant to high-dose cabergoline; a phase III study of lapatinib and cabergoline in resistant prolactinomas is ongoing.
MEDICAL TREATMENT OF NON-FUNCTIONING PITUITARY ADENOMAS
Most non-functioning pituitary adenomas express D2-receptors and tumor growth can be potentially controlled by cabergoline [53]. Study results are controversial and initial studies included non-progressing tumors; therefore, treatment efficacy is difficult to ascertain [5455]. A recent study by Greenman et al. [56] showed a promising response rate of 58% in enlarging tumors; however, the study was limited by its retrospective design, group imbalances, and inherent selection and observation bias. Thus, with the absence of randomized placebo-controlled trials, use of cabergoline in patients with non-operable tumors or with enlarging residual tumors cannot be universally recommended.
MEDICAL TREATMENT OF AGGRESSIVE/ATYPICAL PITUITARY ADENOMAS AND PITUITARY CARCINOMAS
Atypical adenomas are extremely invasive and rapidly progressing pituitary tumors. Characterized by pathologic features such as high Ki-67 (more than 3% to 10%) with extensive positive expression of p53 protein [57], they represent 5% to 10% of pituitary tumors. Temozolomide (TMZ), an oral chemotherapy, used alone [58] or in combination with DAs [59] or SRLs [60] may have an additional effect in tumor control. Data to support TMZ use is scarce; however, it should be considered alongside surgery and radiation therapy in patients with aggressive tumors.
Pituitary carcinomas, defined by presence of widespread metastasis, are exceedingly rare. TMZ response rate in a meta-analysis on pituitary carcinomas was 65% with improved overall 5-year survival of 92% versus 54% (P=0.08) [58]. Response is usually, but not always, inversely proportional to the tumoral expression (immunostaining) of 6-methyguanine-DNA methyltransferase, a DNA repair enzyme that interferes with TMZ action.
CONCLUSIONS
Knowledge of tumor pathophysiology and biology is rapidly increasing and pharmacotherapy for pituitary tumors is evolving. Targeting specific receptors and genes implicated in tumors pathogenesis and determining predictors of response using radiological, pathological, and clinical characteristics will lead to more personalized medicine. It is our hope that care of patients with pituitary tumors will continue to advance over time, achieving both biochemical and tumor response, but also significant improvement in quality of life and comorbidities.
Notes
CONFLICTS OF INTEREST: Maria Fleseriu has been a principal investigator with research grants to OHSU and scientific consultant to Chiasma, Novartis, Pfizer, Strongbridge. Fabienne Langlois and Shirley McCartney have no conflict of interest.