Connecting Myokines and Metabolism
Article information
Abstract
Skeletal muscle is the largest organ of the body in non-obese individuals and is now considered to be an endocrine organ. Hormones (myokines) secreted by skeletal muscle mediate communications between muscle and liver, adipose tissue, brain, and other organs. Myokines affect muscle mass and myofiber switching, and have profound effects on glucose and lipid metabolism and inflammation, thus contributing to energy homeostasis and the pathogenesis of obesity, diabetes, and other diseases. In this review, we summarize recent findings on the biology of myokines and provide an assessment of their potential as therapeutic targets.
INTRODUCTION
Lack of exercise and sedentary lifestyle have been linked to obesity, type 2 diabetes, cardiovascular diseases, cancer, osteoporosis, and premature death [1234567]. Skeletal muscle is the most abundant tissue in non-obese adults, accounting for approximately 40% of the body weight [8]. Skeletal muscle adapts to mechanical, neural and humoral stimuli, and plays critical roles in physical activity, energy expenditure, and glucose disposal [910]. Exercise and anabolic hormones, e.g., insulin, insulin-like growth factor 1, growth hormone and testosterone, increase skeletal muscle mass (Fig. 1) [1112]. Conversely, physical inactivity from aging or neuromuscular disorders, and chronic diseases, such as cancer, renal failure, respiratory failure, infection, and some endocrine disorders, e.g., uncontrolled diabetes mellitus, hyperthyroidism and hypercortisolism, cause muscle atrophy or "sarcopenia" (Fig. 1) [1314151617]. Sarcopenia has been linked to obesity, metabolic syndrome, and other diseases in aged populations, particularly in South Asia (Fig. 2) [18192021].

Positive and negative regulators of skeletal muscle mass. Myokines are produced and secreted by skeletal muscle and act via autocrine, paracrine and endocrine mechanisms to regulate skeletal muscle mass and metabolism. IGF-1, insulin-like growth factor 1; IL, interleukin; BDNF, brain-derived neurotrophic factor; FGF-21, fibroblast growth factor 21; LIF, leukemia inhibitory factor.
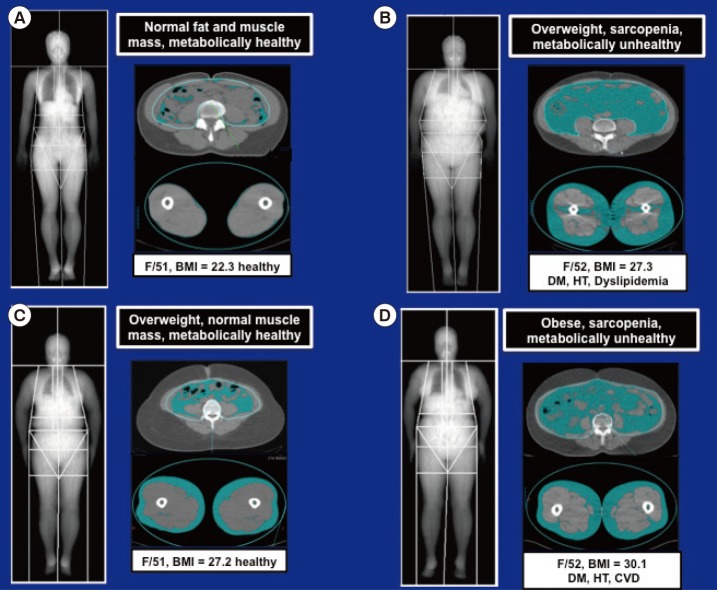
Magnetic resonance imaging scans comparing the distributions of abdominal and thigh fat and muscle in (A) lean, (B, C) overweight, and (D) obese women. As in (B) and (D), increased visceral adiposity and sarcopenia are associated with diabetes mellitus (DM), hypertension (HT), dyslipidemia, and cardiovascular disease (CVD). BMI, body mass index.
The concept that skeletal muscle secretes humoral factors that actively communicate with other organs was proposed many years ago [222324]. Henningsen et al. [25] and Pedersen et al. [26] used the term "myokines" to describe cytokines and other peptides expressed and released by muscle cells. This review highlights the biological actions of myostatin and other myokines that regulate skeletal muscle mass and metabolism via autocrine, paracrine, and endocrine mechanisms.
MYOSTATIN
Much attention has been focused on the biology of the transforming growth factor β (TGF-β) superfamily of proteins since the discovery of myostatin [27]. Myostatin, also known as growth differentiation factor 8, is expressed and secreted predominantly by skeletal muscle and inhibits muscle growth. This function is conserved in many species, as evident by the hypermuscular phenotype resulting from inactivation of myostatin gene in mice, sheep, cattle, and human [2829303132]. During early postnatal development, myostatin inhibits muscle stem cell proliferation, differentiation, and protein synthesis [33]. Normally, the differentiation of skeletal muscle cells requires growth arrest followed by expression of muscle-specific genes. These processes are coordinated by activation of specific cyclins, cyclin-dependent kinases (Cdk), Cdk inhibitors (CdkIs), and muscle regulatory factors. During the proliferation phase, myostatin up-regulates p21 (a CdkI), and decreases the levels of Cdk2 and Cdk4, leading to cell cycle arrest.
Myostatin inhibits satellite cell activation by down-regulating the transcription factor Pax7, and also controls the myogenic differentiation program through inhibition of myogenic regulatory factors, such as Pax3, MyoD, and Myf5. Studies indicate that myostatin's inhibitory effect on muscle differentiation in the postnatal period is mediated partly by perturbation of Akt/mammalian target of rapamycin complex1 signaling [34353637]. In mature adult muscle fibers, the C-terminal dimer of myostatin binds to activin receptors II (ActRII), mainly ActRIIB and to a lesser degree ActRIIA, which then recruits, phosphorylates and activates activin receptor-like kinase (Alk) 4 and Alk5, leading to phosphorylation and activation of Smad2 and Smad3 [3839]. Phosphorylated Smad2 and Smad3 form a heterodimeric complex with Smad4, which is translocated into the nucleus, and acts as a transcription factor to regulate gene expression. Myostatin signaling also leads to activation of Smad7 which functions as a negative feedback inhibitor [4041]. The activation of myostatin-Smad pathway inhibits the translation initiation complex and protein synthesis. Myostatin suppresses Akt signaling and acts via forkhead box protein O1 transcription factors to promote protein breakdown through activation of the ubiquitin-proteasome system (Fig. 3). Myostatin also inhibits the autophagy-lysosome system [4243].
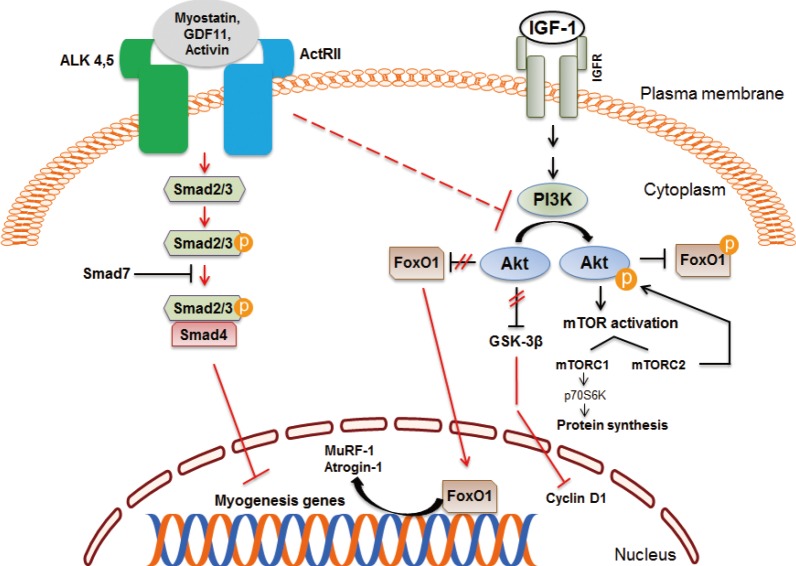
Myostatin and insulin-like growth factor 1 (IGF-1) signaling pathways in skeletal muscle. Myostatin and other transforming growth factor β family members signal via activin receptor II (ActRII), Smad2, and Smad3, which blocks muscle differentiation and leads to muscle atrophy. Inhibition of regulatory-associated protein of mammalian target of rapamycin (mTOR) and mTOR complex 1 (mTORC1) has an additive effect on myostatin signaling. The IGF-1/Akt pathway induces skeletal muscle hypertrophy. ALK, activin receptor-like kinase; GDF11, growth differentiation factor 11; IGFR, IGF receptor; FoxO1, forkhead box protein O1; PI3K, phosphatidylinositol 3-kinase; GSK-3β, glycogen synthase kinase 3β; MuRF, muscle ring finger; p70S6K, p70 S6 kinase.
Genetic or pharmacologic inhibition of myostatin, ActRIIB, Alk4/Alk5, or Smad2/3 results in skeletal muscle hypertrophy, associated with increased protein synthesis and reduced protein degradation [44]. Myostatin knockout (Mstn-/-) mice have increased skeletal muscle mass as well as reduced body fat [45]. Myostatin-null agouti lethal yellow or leptin deficient mice have drastically reduced body fat and glucose levels raising the possibility that blockade of myostatin signaling may be useful for treating obesity and diabetes [46]. Guo et al. [47] have shown that Mstn-/- mice have increased glucose utilization and insulin sensitivity. To determine whether these effects were due to a lack of myostatin signaling in muscle or adipose tissue, they compared the metabolic phenotypes of mice carrying a dominant negative ActRIIB receptor expressed in adipocytes or skeletal muscle. The absence of myostatin signaling in adipocytes did not affect body composition or glucose homeostasis, whereas inhibition of myostatin signaling in skeletal muscle recapitulated the phenotype of Mstn-/- mice, characterized by hypermuscularity, decreased body fat, and enhanced insulin sensitivity [47].
We studied the effects of pharmacological blockade of myostatin and related peptides by treating mice on chow and high-fat diets with a soluble activin receptor type IIB (ActRIIB-Fc). ActRIIB-Fc treatment increased lean and skeletal muscle mass, grip strength, and contractile force, decreased body fat, and increased insulin sensitivity [48]. Mice lacking Akt1 or Akt2 have reduced muscle mass, grip strength and contractile force, consistent with a pivotal role of Akt signaling in promoting muscle growth [4950]. Contrary to in vitro studies showing that Akt signaling is necessary for the ability of ActRIIB inhibition to induce muscle hypertrophy, we found that Akt1 and Akt2 deficient mice responded similarly as wild type mice to ActRIIB-Fc in regard to increased muscle size, grip strength and contractile force, indicating these Akt isoforms are not essential for ActRIIB signaling [51].
ActRIIB-Fc has also been shown to decrease diet-induced obesity and improve glucose and lipid levels in mice [48]. Importantly, ActRIIB-Fc induced the browning of white adipose tissue (WAT), as shown by increased expression of the thermogenic genes uncoupling protein 1 (UCP1) and peroxisomal proliferator-activated receptor-γ coactivator 1α (PGC1α). Thus, the anti-obesity effect of ActRIIB-Fc is partly by increasing skeletal muscle mass as well as inducing thermogenesis in WAT [52]. Other studies have confirmed that deficiency of myostatin signaling in Mstn-/- mice promotes browning of WAT [5354]. WAT of Mstn-/- mice displays features of brown adipose tissue, e.g., increased expression of including UCP1 and PGC1α, as well as expression of beige adipocyte markers, e.g., Tmem26 and CD137. The enhanced browning of adipose tissue appears to be mediated by irisin (fibronectin type III domain-containing 5, Fndc5), a myokine secreted from skeletal muscle in Mstn-/- mice. Myostatin deficiency stimulates AMPK expression and phosphorylation, which then activates PGC1α and irisin and promotes the browning of adipose tissue and thermogenesis [54]. Another study has shown that the reduction of body fat in Mstn-/- mice is due to increased energy expenditure and leptin sensitivity [55]. The cross-talk of myokines and adipokines may provide novel therapeutic tools for treating obesity, diabetes, and diseases associated with muscle atrophy.
Does myostatin blockade have clinical potential? A double-blind, placebo-controlled study evaluated the safety, pharmacokinetics, and pharmacodynamics of a decoy ActRIIB receptor (ACE-031) in healthy postmenopausal women randomized to receive a single dose of ACE-031 (0.02 to 3 mg/kg subcutaneous) or placebo. ACE-031 treatment had mild adverse events and produced significant increases of lean mass and thigh muscle volume at day 29 in those receiving 3 mg/kg. Moreover, ACE-031 treatment increased adiponectin by 51.3% and decreased leptin by 27.7% demonstrating a favorable metabolic profile [56]. Androgen deprivation therapy for prostate cancer causes sarcopenia and increased body fat. An anti-myostatin peptibody (AMG 745/Mu-S) was evaluated in men undergoing androgen deprivation therapy for non-metastatic prostate cancer [57]. The adverse events in AMG 745 versus placebo treated groups were: diarrhea (13% vs. 9%), fatigue (13% vs. 4%), contusion (10% vs. 0%), and injection site bruising (6% vs. 4%). AMG 745 treatment increased the lean body mass and decreased fat mass. These preliminary results provide support for further investigation into the safety profile and of therapeutic uses of myostatin blockade to reduce sarcopenia and improve metabolism.
As discussed earlier, myostatin deficiency or blockade of ActRIIB receptor potently decreases body fat and improves metabolic outcomes in obese mice [535455]. Human obesity is associated with increased myostatin expression and plasma myostatin levels. The secretion of myostatin from myotubes derived from muscle biopsies is increased in obese compared with lean women [5859]. The biological significance of these findings, and whether myostatin and other TGF-β peptide superfamily can be targeted specifically for treatment of obesity and metabolic disorders requires further studies.
INTERLEUKIN 6
The cytokine interleukin 6 (IL-6) was named a myokine because its levels increased in response to exercise and muscle contraction [606162]. Evidence supporting the notion that is the source of IL-6 is based on transcriptional analysis of IL-6 mRNA levels during exercise, in situ hybridization and immunohistochemistry of IL-6, microdialysis of contracting skeletal muscle, and measurement of arteriovenous IL-6 concentrations and blood flow across an exercising leg [63]. Skeletal muscle adapts to exercise by altering glycogen content, increasing β-oxidation of fatty acids, increasing intramyocellular triglyceride hydrolysis, and enhancing epinephrine-induced lipolysis [64]. Thus, the trained skeletal muscle uses fat as a substrate and is less dependent on glucose and muscle glycogen during exercise.
Epidemiological studies have found an inverse correlation of the amount of physical activity and plasma IL-6 concentration. The basal plasma levels of IL-6 are strongly associated with physical inactivity, obesity and metabolic syndrome [656667]. Chronic exercise decreases the basal levels of IL-6, and the increases in plasma IL-6 and muscle IL-6 mRNA content during acute exercise are also blunted in response to endurance training [68]. IL-6 receptor (IL-6R) α is regulated opposite to IL-6, and the basal IL-6Rα mRNA content in muscle is increased during endurance training, perhaps counteracting the reduction in IL-6 [69].
What are the biological roles of IL-6? Treatment of rat L6 myocytes with IL-6 increases basal glucose uptake via glucose transporter 4 translocation, as well as insulin-stimulated glucose uptake [70]. The in vitro effect of IL-6 on glucose uptake is mediated, at least partly, through AMP-activated protein kinase (AMPK) activation. Studies have also suggested that IL-6 may stimulate fatty acid oxidation via AMPK [717273]. In resting humans, acute administration of IL-6 infused to achieve physiological concentrations had no effect on either endogenous glucose production or glucose disposal [74]. In contrast, when IL-6 was infused to mimic the plasma of IL-6 observed during high-intensity exercise, the endogenous glucose production was markedly increased, suggesting that a muscle-liver crosstalk mediated via IL-6 may have a role in regulating plasma glucose levels through endogenous glucose production during exercise [70]. In addition to its effects on glucose metabolism, infusion of IL-6 in healthy volunteers stimulates lipolysis in skeletal muscle without affecting adipose tissue [7175]. IL-6 inhibits endotoxin-induced tumor necrosis factor (TNF) production in human monocytes, and infusion of IL-6 during exercise attenuates the ability of endotoxin to increase TNF levels in healthy individuals. These anti-inflammatory properties of IL-6 are associated with induction of anti-inflammatory cytokines, e.g., IL-1 receptor agonist and IL-10 [76].
INTERLEUKIN 15
IL-15 was classified as an interleukin based on its 4-α-helical secondary structure and its ability to mimic the functions of IL-2 [77]. The plasma membrane receptor for IL-15 was shown to be composed of IL-2 receptor β (IL-2Rβ), the common gamma chain (γc), and a specific IL-15 receptor α (IL-15Rα) chain [7879]. Transcripts for IL-15 and IL-15Rα are widely expressed, and skeletal muscle expresses IL15 and IL15RA mRNAs [7778]. In vitro experiments in myogenic cells suggested that IL-15 was an anabolic factor for skeletal muscle; however, increasing IL-15 levels in vivo did not induce muscle hypertrophy [80818283]. Nonetheless, studies have revealed different locomotor phenotypes of mice lacking IL-15Rα or IL-15 or IL-2Rβ [8485]. Furthermore, single nucleotide polymorphisms (SNPs) in the human IL15 and IL15RA genes have been associated with different muscle phenotypes, responses to resistance training, and obesity [8687888990].
We hypothesized that IL-15Rα has a role in determining the muscle phenotype in mice. We found that loss of IL-15Rα leads to a remodeling of fast skeletal muscles to a more oxidative phenotype associated with increased spontaneous locomotor activity and exercise capacity, and resistance to fatigue [91]. The molecular signature of oxidative muscle phenotype from IL-15Rα knockout mice included altered mitochondrial biogenesis and calcium homeostasis. Consistent with our observations in mice, we found a significant association between a SNP in exon 3 of the IL15RA gene and endurance in human athletes [91].
A recent paper by O'Connell and Pistilli [92] has shown mechanisms by which IL-15Rα induced an oxidative skeletal muscle phenotype. Muscle-specific deletion of IL-15Rα resulted in a greater mitochondrial density and reduced twitch:tetanus ratio in extensor digitorum longus and soleus muscles indicating a oxidative shift in muscle phenotype. However, the spontaneous activity was not different in muscle IL-15Rα deficient mice, unlike the whole body IL-15Rα knockout mouse [93]. Thus, muscle IL-15Rα has a role in altering contractile properties and fatigue characteristics of skeletal muscles, but the locomotor behavior is likely to be controlled by IL-15Rα targets in brain [84]. Further studies are needed to evaluate whether IL-15Rα can be targeted specifically for obesity treatment by increasing energy expenditure and fatty acid oxidation.
IRISIN
Chronic exercise increases skeletal muscle mitochondrial biogenesis, which is regulated by PGC1α [949596]. Bostrom et al. [97] demonstrated that the inguinal subcutaneous WAT had increased levels of UCP1 and Cidea in muscle-specific PGC1α transgenic mice compared to wild-type mice. To address whether the browning of the subcutaneous WAT was due to a myokine, they cultured primary murine subcutaneous adipocytes with conditioned media from PGC1α overexpressing myocytes, and found that the conditioned media increased expression of brown-fat specific genes in adipocytes. Using gene array and bioinformatic methods, Bostrom et al. [97] identified FNDC5 as a gene target of PGC1α, and showed that FNDC5 expression was increased in muscle obtained from exercise-trained mice and humans. Primary subcutaneous adipocytes treated with recombinant-FNDC5 displayed an increased expression of brown adipose genes, i.e., UCP1, Elovl3, Cox7a, and Otop1. Moreover, UCP1-positive cells treated with FNDC5 developed multi-loculated lipid droplets, increased mitochondrial content and oxygen consumption, consistent with a thermogenic phenotype. Based on these results, the authors surmised that FNDC5 induced a beige phenotype of WAT in mice, and this effect was attenuated by a peroxisome proliferator-activated receptor α antagonist treatment [97]. Further experiments revealed that the full-length FNDC5 was a transmembrane protein, and the extracellular N-terminal portion of FNDC5 was secreted and was highly homologous between mouse and humans. This myokine was named "irisin" after the Greek messenger goddess Iris. Plasma irisin levels were shown to be increased in mice and humans after short-term exercise. Adenoviral expression of FNDC5 in liver increased plasma irisin levels which led to the browning of subcutaneous WAT, increased energy expenditure, and protection against obesity and insulin resistance [97].
However, questions have been raised about the biology of FNDC5 expression and plasma irisin levels [9899]. It is unclear whether irisin is truly a myokine since human WAT is capable of expressing FNDC5 and secreting irisin [100]. Some studies indicate that neither acute nor chronic exercise consistently increases expression FNDC5 and/or irisin in humans [101102]. However, others have shown associations of plasma irisin and aging, obesity, physical activity, and metabolic outcomes [103104]. These controversies surrounding the role of irisin may arise from different exercise regimens and assays for measuring irisin, suboptimal storage of tissue samples, as well as differences in the function of irisin in mouse versus human [101104105].
CONCLUSIONS
Myokines are proposed to play important roles in mediating the beneficial effects of skeletal muscle mass and exercise on health. Myokines have been implicated in the pathogenesis of obesity, substrate oxidation, lipid partitioning, insulin sensitivity, and inflammation. While the list of putative myokines keeps growing, the specific physiological and pathological effects of these molecules are poorly understood. Important questions that need to be answered for a presumed myokine include whether skeletal muscle is the main or only source, how the local and systemic concentrations of the myokine are regulated, whether there are biological differences among species, and what specific signaling mechanisms mediate the biological effects of the myokine in various organs. A better understanding of the actions of myokines may identify novel therapies for obesity, diabetes, cardiovascular diseases, cancer, and other diseases known to be improved by exercise.
ACKNOWLEDGMENTS
We thank Dr. Lim Soo of the Seoul National University College of Medicine for providing MRI scans (Fig. 2). RSA is supported by American Diabetes Association grant #7-13-BS-004, and National Institutes of Health grants R01-NS084965 and P01-DK049210.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.